- Search Menu
- Sign in through your institution
- Advance Articles
- Editor's Choice
- Information for authors
- Submission Site
- Open Access Options
- Why publish with the journal
- About DNA Research
- About the Kazusa DNA Research Institute
- Editorial Board
- Advertising and Corporate Services
- Journals Career Network
- Self-Archiving Policy
- Dispatch Dates
- Journals on Oxford Academic
- Books on Oxford Academic

Browse issues

Cover image

Volume 31, Issue 6, December 2024
Resource article: genomes explored, chromosome-scale genome assembly of acerola ( malpighia emarginata dc.).
- View article
- Supplementary data
Chromosome-level genome assembly of the medicinal insect Blaps rhynchopetera using Nanopore and Hi-C technologies
Research articles, the burst of satellite dna in leptidea wood white butterflies and their putative role in karyotype evolution, time-dependent changes in genome-wide gene expression and post-transcriptional regulation across the post-death process in silkworm, email alerts.
- Author Guidelines
- X (formerly Twitter)
Affiliations
- Online ISSN 1756-1663
- Copyright © 2024 Kazusa DNA Research Institute
- About Oxford Academic
- Publish journals with us
- University press partners
- What we publish
- New features
- Open access
- Institutional account management
- Rights and permissions
- Get help with access
- Accessibility
- Advertising
- Media enquiries
- Oxford University Press
- Oxford Languages
- University of Oxford
Oxford University Press is a department of the University of Oxford. It furthers the University's objective of excellence in research, scholarship, and education by publishing worldwide
- Copyright © 2024 Oxford University Press
- Cookie settings
- Cookie policy
- Privacy policy
- Legal notice
This Feature Is Available To Subscribers Only
Sign In or Create an Account
This PDF is available to Subscribers Only
For full access to this pdf, sign in to an existing account, or purchase an annual subscription.
Thank you for visiting nature.com. You are using a browser version with limited support for CSS. To obtain the best experience, we recommend you use a more up to date browser (or turn off compatibility mode in Internet Explorer). In the meantime, to ensure continued support, we are displaying the site without styles and JavaScript.
- View all journals
- Explore content
- About the journal
- Publish with us
- Sign up for alerts
- Perspective
- Published: 25 November 2024
The lives of cells, recorded
- Amjad Askary ORCID: orcid.org/0000-0002-2913-8498 1 na1 ,
- Wei Chen ORCID: orcid.org/0000-0002-5255-4166 2 , 3 na1 ,
- Junhong Choi ORCID: orcid.org/0000-0001-9291-5977 2 , 4 na1 ,
- Lucia Y. Du ORCID: orcid.org/0000-0003-0151-3783 5 , 6 na1 ,
- Michael B. Elowitz ORCID: orcid.org/0000-0002-1221-0967 6 , 7 , 8 ,
- James A. Gagnon ORCID: orcid.org/0000-0003-3978-6058 9 ,
- Alexander F. Schier ORCID: orcid.org/0000-0001-7645-5325 5 , 6 ,
- Sophie Seidel ORCID: orcid.org/0000-0002-4484-9888 10 , 11 na1 ,
- Jay Shendure ORCID: orcid.org/0000-0002-1516-1865 2 , 6 , 12 , 13 , 14 ,
- Tanja Stadler ORCID: orcid.org/0000-0001-6431-535X 10 , 11 &
- Martin Tran ORCID: orcid.org/0000-0001-9882-7230 7 na1
Nature Reviews Genetics ( 2024 ) Cite this article
7251 Accesses
161 Altmetric
Metrics details
- CRISPR-Cas systems
- Developmental biology
- Genetic engineering
- Phylogenetics
A paradigm for biology is emerging in which cells can be genetically programmed to write their histories into their own genomes. These records can subsequently be read, and the cellular histories reconstructed, which for each cell could include a record of its lineage relationships, extrinsic influences, internal states and physical locations, over time. DNA recording has the potential to transform the way that we study developmental and disease processes. Recent advances in genome engineering are driving the development of systems for DNA recording, and meanwhile single-cell and spatial omics technologies increasingly enable the recovery of the recorded information. Combined with advances in computational and phylogenetic inference algorithms, the DNA recording paradigm is beginning to bear fruit. In this Perspective, we explore the rationale and technical basis of DNA recording, what aspects of cellular biology might be recorded and how, and the types of discovery that we anticipate this paradigm will enable.
You have full access to this article via your institution.
Similar content being viewed by others

Lineage tracing and analog recording in mammalian cells by single-site DNA writing

Molecular recording using DNA Typewriter

A time-resolved, multi-symbol molecular recorder via sequential genome editing
Introduction.
A fundamental challenge in biology is to explain biological states or processes, including normal development and disease, in terms of the events that precede them. The sequence of events taking place in individual cells can be collectively thought of as a cellular history . Cellular histories include a range of features about each cell that encapsulate past relationships and events, inform its present state, and constrain its possible futures (Fig. 1a ), including:
How a cell is related to other cells by lineage, that is, which cells are its sister, its cousins and so forth. Cellular phenotypes, functions and potentials are often inherited through, or structured by, mitotic divisions 1 .
The identity, amplitude and duration of extrinsic signals (mechanical or biochemical) that a cell, or its ancestors, received at different points in its past. Such signals provide cues for changes to cell state or cell fate 2 , or responses to environmental stresses or injuries.
The longitudinal dynamics of each cell’s internal molecular state (that is, its trajectory ), including the levels of transcription factors and other regulatory molecules, as well as its epigenome. These characteristics constrain and often determine a cell’s functional behaviours, including its ability to change into other states 3 .
Spatial context, including the identities and states of a cell’s past and present neighbours. The spatial neighbourhood of a cell informs its function within tissue and reflects its developmental history of growth, differentiation and cell movement 4 .

a , Longitudinal recordings of cellular histories are needed to understand their dynamics, such as cell lineage (1), extrinsic signalling (2), internal state (3) and spatial relationships (4) that give rise to cell states and fates over time. b , Recording in diverse biological contexts may enable insights into the origin(s) of adult cell types, the evolution of clonality, the intrinsic and extrinsic signalling history associated with final cell fate, and microenvironments. c , A decorated tree reconstructed from DNA recordings provides multimodal cell histories (see panel a ) over time.
Cellular histories have the potential to show not only how individual cells are related to one another, but also what events drove them into their observed states, in diverse biological contexts (Fig. 1b ). For example, in developmental biology, cellular histories can help to delineate the developmental potentials of all progenitors, and can conclusively determine the origin(s) of both healthy and disease-associated adult cell types 5 , 6 , 7 . Applied to cancer progression, cellular histories reveal how clonal fitness evolves in tumours, yielding reconstructions of the emergence of invasiveness as well as the spatiotemporal dynamics of metastasis 8 . Reconstructing the intrinsic and extrinsic signalling history of individual cells can inform our understanding of cell fate determination, and of spatial and temporal patterning of tissues and organs during development 9 , 10 , 11 . In microbial environments, such as soil or the human body, cellular histories can reveal how microorganisms respond to different environmental cues and conditions 12 .
From a technical standpoint, cellular histories are challenging to obtain. The most obvious approach is through direct visualization of cells over time. However, many model organisms and microbial environments are composed of millions to trillions of cells generated through variable patterns of cell division and traversing non-deterministic trajectories. In addition to scalability limitations, the direct observation of cells is complicated by the opaqueness of organisms, their physical movement during activities of interest, and the long timescales over which biology plays out. An alternative to visualization involves genome-wide methods for measuring cell state, such as multiplex fluorescence in situ hybridization (FISH) or single-cell RNA sequencing (scRNA-seq). For example, the widespread application of scRNA-seq has revealed a myriad of cell types and cell states in diverse organisms 13 , 14 , 15 . However, these approaches are typically destructive, preventing continuous monitoring of the same cells over time. Since trajectories often involve reversible or discontinuous changes, it is difficult to reliably infer a series of states traversed by individual cells from a series of snapshots 16 .
Building on a rich history (Box 1 ), the nascent field of DNA-based recording offers a potential way of obtaining cellular histories that overcomes the limitations of current measurement paradigms. The basic premise of DNA-based recording is to engineer cells to record their histories into their genomes using DNA editors, such that a single destructive snapshot is informative with respect to not only each cell’s present but also its past. A key principle that enables DNA-based recording is the generation of heritable mutations at defined target sites. Two types of system can be used to generate edits: constitutive recorders generate random edits on a set of target sites, allowing reconstruction of lineage trees 9 , 17 , 18 , whereas conditional recorders generate edits at a different set of target sites at a rate proportional to the external signals, internal states or spatial contexts being sensed, allowing analysis of the past trajectory of single cells 17 , 19 . Many conditional editors and corresponding target sites can be included to record as many biological inputs of interest as needed. When both systems are activated, the result is a cell lineage tree ‘ decorated ’ with the relative activity of specific biological events over cell divisions or absolute time (Fig. 1c ) across different biological contexts for which recording has strong potential to provide insight.
This Perspective focuses on the present and future of DNA-based recording, highlighting recent technological advances. Breakthroughs in DNA synthesis 20 , assembly 21 , 22 and delivery 23 are facilitating the engineering of genomes to encode the necessary components of read/write systems. Site-specific recombinases 24 , clustered regularly interspaced short palindrome repeats (CRISPR) systems 25 , 26 , 27 and other genome-editing technologies 28 are enabling the writing of information to the genome, while advances in single-cell molecular profiling, spatial transcriptomics and sequencing are enabling its recovery 29 , 30 . Finally, progress in big data analysis and phylogenetic inference algorithms is furthering our ability to reconstruct complex cellular histories from DNA edits 31 . Of note, although RNA-based 32 , 33 , 34 and protein-based 35 , 36 recording methods have been described recently, we focus here on recording to genomic DNA, which allows long-term storage and faithful transmission of recordings through not only cell divisions but also throughout the life of an organism. For practical guidance on how recording experiments can be designed, executed and interpreted, we refer the reader to other recent reviews 16 , 37 , 38 , 39 .
Box 1 A historical perspective on recording cell histories
Over the past two centuries, various modes for recording cell histories have emerged that can be broadly categorized into: lineage tree reconstruction, where each branch ideally represents single cell divisions; clonal analysis, where cell fates that descended from a common progenitor are grouped together; and signal recording, where previously experienced signals provide an indelible mark within cells.
Strategy 1: Reconstructing trees of embryonic development through direct observation
The field of cell lineage tracing has deep roots that extend to the late nineteenth century, when Charles Whitman, Edwin Conklin and Edmund Wilson independently observed cells in embryos to trace the developmental origins of germ layers in various marine invertebrates 145 , 146 , 147 . Strikingly, they found that these embryonic cell divisions often occur in a stereotypical, invariant manner that was similar between different species. Conklin began constructing larger fate maps 148 — schematics that delineate the future fates of individual progenitor cells or regions. Decades later, John Sulston and colleagues used direct observation to map the complete lineage tree that gives rise to the cells of the hatched larvae of Caenorhabditis elegans 149 . These studies demonstrated how relatively simple observational methods can be used to construct detailed maps of embryonic development. However, tissues and embryos in many organisms are not transparent, are larger in scale and proceed more slowly and less deterministically in their development than in C. elegans , precluding the use of direct observation to reconstruct complete cell lineages.
Strategy 2: Clonal analysis through dye injections, transplantation, chimaera generation, retroviral infection and in vitro culture systems
Experimental manipulations can be used to trace the contributions of progenitor cells to descendants. For example, researchers began using dyes and radioactive tracers to label groups of progenitor cells 150 , 151 , 152 , transplanting tissue to foreign hosts 153 and generating chimaeras through embryo aggregation 154 , 155 . These approaches produced fundamental discoveries about fate bias and cell migration. For example, the construction of fate maps within amphibian embryos revealed that vertebrate embryos are generally divided into three germ layers, which contain progenitors that give rise to particular sets of future organs. Later approaches permanently labelled single cells by reporter gene transfection to analyse the fates of their progeny 156 , 157 , 158 , 159 . In vitro culture systems were also used to track individual progenitors using time-lapse microscopy 160 . These enhanced versions of direct observation revealed that clones could be surprisingly variable in both size and descendant-cell diversity. Unlike C. elegans development, vertebrate development is much more plastic. However, although these approaches are powerful, portable and still widely used, they are best suited to looking at a single process or cell type at a time, and are difficult to scale to all cells in a tissue or organism.
Strategy 3: Clonal analysis in specific subsets of progenitors using genetic recombinases
Genetic recombinases began to be used widely in the 1990s to analyse groups of progenitors marked by the expression of key genes 161 , 162 , 163 . These experiments often involved expressing a recombinase (Cre or FLP) in a cell-type-specific or tissue-specific manner to permanently activate a reporter gene to label all descendant cells. These studies generally revealed that while some progenitor cells seem to be multipotent, giving rise to many fates, other progenitor cells can have distinct and reproducible biases in cell fate 164 , 165 , 166 . Nevertheless, this work was limited to analysing groups of progenitor cells, since all activated progenitors would be identically labelled, preventing high-resolution, single-cell analyses of lineage and fate. More complex recombination cassettes were later developed to label different cells with distinct colours or combinations of colours 42 , 43 , 167 , 168 . These allowed the simultaneous analysis of dozens of clones within a tissue, providing insight into mechanisms like clonal crypt formation in the intestine and subsets of neural progenitors in the developing brain. However, these methods were not scalable to larger tissues or organs, because limited colour diversity meant that unrelated cells could be coincidentally labelled with the same colour combination with high likelihood.
Genetic recombinases have also been adapted for recording developmental signalling pathway activation and neural activity. These approaches rely on signalling-pathway-specific promoters that express a recombinase, often gated by small-molecule activation enabling temporal control of recording, leading to permanent activation of a reporter gene. Importantly, these approaches typically record the transcriptional activation of a signalling pathway, not the expression of a signalling molecule itself. Treating embryos with a drug during a window of developmental time will permanently label all cells that activate a given signalling pathway. This approach has been used, for example, in mouse embryos to link Shh signalling to limb and digit patterning 169 , 170 and forebrain neurogenesis 171 . More recently, genetic recombinases were used in adult animals to label and manipulate the neural circuits that control cognition and behaviour 172 , 173 . Current strategies label specific subsets of neurons for manipulation based on immediate early gene expression, intracellular calcium levels 174 , 175 or synaptic activity 176 , 177 . These approaches typically also suffer from the inability to mark different cells with distinguishable labels.
Strategy 4. Reconstructing lineage trees using naturally occurring somatic mutations
In most approaches discussed thus far, labels are experimentally delivered to cells that subsequently proliferate and differentiate. These prospective approaches are limited to experimentally accessible systems and cannot be applied to the study of human embryo development. As an alternative, somatic mutations that accumulate naturally due to errors in DNA replication can be sequenced and used to reconstruct lineage trees retrospectively. As a proof of principle, researchers began to use sequencing to trace lymphoid lineages through somatic hypermutation at immunoglobulin loci 178 , intestinal lineages through post-zygotic methylation patterns at CpG sites 179 , and epithelial lineages through polyguanine repeat DNA sequences 180 . With new innovations in single-cell DNA and RNA sequencing in the 2010s, somatic mutations could be detected at scale and used for lineage reconstruction, including single-nucleotide variants 181 , 182 , 183 , 184 , 185 , copy-number variants 186 and microsatellite repeats 187 . These approaches have been used to study human embryogenesis, revealing a surprisingly unequal contribution of early embryonic cells to adult somatic tissues 188 . However, although powerful, these approaches are only able to reconstruct lineage trees, because the transient trajectories that influence human cell-type differentiation are not recorded in DNA-replication errors.
Strategy 5: High-throughput clonal analysis by sequencing static barcodes
In 1992, Walsh and Cepko pioneered the use of diverse retrovirus libraries containing short unique DNA sequences to distinguish between individual clones within the same tissue at scale 189 . Initially, this approach was laborious because individual cells had to be excised and processed to enable PCR to detect the barcode. However, scalable single-cell sequencing in the 2010s opened up the era of single-cell clonal barcoding, allowing the simultaneous analysis of many clones containing endpoint cell states. Multiple approaches for clonal barcoding, such as diverse viral libraries, multiplexed recombination cassettes or mobile transposable elements, were first applied to the study of haematopoiesis. These studies revealed migration patterns of antigen-specific T cells 190 , fate potential in lymphoid-primed multipotent progenitors 191 and hematopoietic stem cell (HSC) progenitors 43 , 192 , and differences in HSC proliferation between irradiated transplantation settings 193 versus steady-state haematopoiesis 194 . Clonal barcoding was later applied to other systems to study fate differentiation in cancer 195 and forebrain development 196 , 197 . Nevertheless, the static nature of these barcodes limited the inferences of temporal dynamics. This limitation has been partially addressed by taking advantage of systems where cells can be grown in vitro. For example, Weinreb et al. 198 barcoded a pool of haematopoietic progenitors, allowed them to expand, then split them to simultaneously profile cell states immediately and at later time points after differentiation, thus linking early transcriptional states to later clonal fate compositions. Biddy et al. 199 applied lentiviral transduction successively to fibroblasts undergoing reprogramming for coarse-scale lineage tree reconstruction, in which branches represent multiple cell divisions. However, these approaches are difficult or impossible to implement in vivo, necessitating other approaches for resolving temporal dynamics.
Recording cellular histories
DNA recording systems integrate recent advances in three areas: (1) writing information through time as heritable DNA edits, either by constitutive or conditional editing; (2) reading recorded information from each cell at an endpoint; and (3) reconstructing the histories of cells from recovered edits (Fig. 2a ).

a , The general DNA recording workflow consists of the following steps: writing information to encode biological information, reading recorded information and reconstructing cell histories. b , Writing information can be done by rearranging, scarring, modifying or inserting DNA. c , Reading recorded information by sequencing or imaging. For sequencing, mRNA encoding barcodes are labelled with unique molecular identifiers and cell barcodes. For imaging, primary probes that bind to the edited and unedited version of the barcode are hybridized — the probe with sequence match outcompetes the one with mismatch for binding. d , Reconstructing trees through DNA recordings allows estimation of the tree topology, branch timescales, ancestral node states and cell dynamics. Edits in the constitutive channel enable tree reconstruction, while edits in the conditional channel allow ‘decoration’ of the tree with experienced signals. cDNA, complementary DNA; CRISPR, clustered regularly interspaced short palindrome repeats; gRNA, guide RNA; pA, polyA; pegRNA, prime editing guide RNA; RT, reverse transcription; T7, T7 RNA polymerase; UMI, unique molecular identifier. Part b adapted with permission from ref. 10 , AAAS.
Writing to cellular histories to DNA
Systems for writing information to the genome would ideally have several characteristics. Writing should be minimally perturbative, that is, neither the editor nor the editing activity should alter cell identity or fate. Recording loci should contain as many target sites as possible to maximize information capacity. Constitutive recorders should be active at a rate that is steady enough and high enough to generate unique edits during each cell cycle. Conditional recorders should be activated by a particular internal or external signal, and be sensitive, selective and quantitative, that is, active in proportion to the biological signals of interest that they sense (for example, enhancer activity, signal duration or protein levels), and inactive in the absence of that signal. Recorders should continue to operate in post-mitotic cells. Each target site should be editable to many distinct and terminal ‘character states’ without the risk of target-site loss. Finally, the sequential order of edits should ideally be recorded to explicitly capture the temporal order of cellular events.
Several classes of editor are being explored as writers, which act by rearranging, scarring, base editing or inserting into genomic DNA. Each has its own strengths and weaknesses (Fig. 2b ).
Rearranging generates edit-state complexity without DNA cleavage
Enhancer-driven site-specific recombinases (SSRs) and integrases have long been used to catalyse programmable rearrangement of DNA sequences as a form of biological recording, for example, recording chemical exposures to population of cells 40 , 41 or permanently marking a subpopulation of cells and its descendants with a fluorescent reporter (Box 1 ). Newer SSR-based recording systems such as intMEMOIR leverage more complex target sites, such as arrays of target sites that stochastically or iteratively rearrange to multiple character states 10 , 42 , 43 . Editing by SSRs avoids endonucleolytic cleavage of DNA strands and does not involve the endogenous DNA repair machinery 44 , 45 . Although the expression of an SSR can be made signal-dependent (for example, by placing it under the control of a cis- regulatory element that is activated by a transcription factor, drug or morphogen), the potential of SSRs to record multiple signals is limited at present by the relatively small number of well characterized, orthogonal SSRs.
Scarring efficiently generates diverse mutations at target edit sites
CRISPR–Cas9 editors can be used to induce DNA double-strand breaks (DSBs) at target sites specified by guide RNAs (gRNAs) 9 , 17 , 18 , 46 , 47 , 48 , 49 . Imperfect repair of those DSBs generates irreversible insertions and deletions (indels), termed ‘scars’. This approach was taken by early attempts at DNA-based lineage recording, such as GESTALT 9 , 50 , and is the most widely applied to date. It has been successful for in vivo recording applications thanks to its high editing rates and capacity for multiplexing with different gRNAs and CRISPR systems 9 , 18 , 46 , 47 , 48 , 49 , 51 , 52 . Furthermore, some in vivo scarring-based implementations place the Cas9 editor under the control of an inducible promoter, that is, single-channel conditional recording 29 . However, DSBs can be toxic to cells, and in arrays of target sites, deletion scars frequently compromise adjacent target sites 9 , 50 , 53 . Moreover, this approach does not directly capture the order in which recorded events occurred (that is, phylogenetic inference across a population of cells is required), although variant approaches based on self-targeting gRNAs can partly mitigate this issue 54 , 55 .
Base editing allows digital editing at precise, densely encoded target sites
CRISPR base editors can generate specific point mutations at target sites 28 , 56 , 57 and have been used for DNA recording 30 , 58 , 59 . Base editors induce mutations without generating DSBs, and allow dense packing of predictably editable target sites into arrays, facilitating subsequent analysis of edit patterns. Although base editors typically generate only one character state, they can be used to produce multiple edit outcomes in two ways. First, newer base editors (such as AXBE and AYBE) allow a single initial base to be edited to all three other bases 60 , 61 . Second, by editing dinucleotides, rather than single-base target sites, it is possible to generate three alternative dinucleotides 62 , 63 . As with indel and rearranging methods, base editing in general produces unordered edits unless the sites are engineered to be edited sequentially 64 , 65 , and can exhibit substantial off-target activity 66 , 67 , 68 , 69 , although this can be in part addressed using newer-generation base editors with less off-target activity 70 , 71 , 72 , 73 .
Inserting allows linearly ordered recording of multiple signal-specific symbols
Prime editors provide a way to generate temporally ordered edits. They are composed of a Cas9 nickase fused to a reverse transcriptase, and use corresponding prime editing guide RNAs (pegRNAs) to insert short sequences precisely at target sites 28 , 74 , 75 . The DNA Typewriter and peCHYRON techniques leverage prime editors to achieve a high number of potential character states for each edit (at least dozens but potentially thousands, if that many pegRNA-expressing constructs could be concurrently introduced) 76 , 77 . Furthermore, in both systems, each insertional edit creates a target site for a new edit. This allows edits to be concatenated sequentially, such that their temporal order is reflected in their linear order along the DNA. Furthermore, prime editors can be used to record transcriptional events by making the production of specific pegRNAs conditional on the activity of a cis -regulatory element, using a framework termed ENGRAM 78 . Similarly, pegRNAs can be activated through protein–protein interactions, a second kind of biologically conditional editing 79 . A drawback with prime editors is that they are currently less efficient than other editors, reducing overall edit rates and temporal resolution, although this may be addressed by newer prime editors 80 .
A second class of insertional editors leverages Cas1–Cas2 systems to integrate short DNA segments (spacers) generated from reverse transcription of intracellular RNA in CRISPR spacer arrays, providing an explicit and ordered record of a cell’s gene-expression history 12 , 19 , 81 , 82 , 83 . With this approach, the acquisition of spacers has been shown to be both orthogonal and dose-dependent on absolute mRNA present within the cell 12 , 82 , 83 . However, these systems are currently limited to prokaryotic systems owing to reliance on accessory integration host factors.
Overall, writers that can deliver precise, information-rich and temporally ordered edits at high rates, without loss of previous recorded information and without perturbing cellular or organismal physiology, are necessary for this paradigm to reach its full potential. At present, prime editors could meet these criteria, especially if they can be optimized to achieve higher edit rates and to perform efficiently across diverse cell types. Base editors could also meet these criteria if they can be optimized to store more memory per edit site and to perform temporally ordered edits in a scalable manner. Cas1–Cas2 systems have the greatest capacity for capturing rich transcriptomic information over time but might require considerable optimization to be successfully ported to eukaryotic systems. Finally, most writers discussed so far have been constitutively expressed to record lineage, and the development of biologically conditional editors remains immature (see section ‘Practical challenges for DNA-based recording’ for further discussion of the challenges).
Reading DNA-recorded information
Information recorded to genomic DNA must eventually be recovered by sequencing or imaging methods. Some approaches directly recover recording information from DNA 9 , 18 , 43 , 58 , but most approaches transcribe the single-copy genomic DNA records into RNA for capture alongside the transcriptome and/or epigenome 29 , 47 , 63 , 84 (Fig. 2c ).
scRNA-seq on dissociated tissues is a convenient and scalable means of obtaining rich endpoint measurements that can also be used to recover DNA-based records. For example, target sites for editors can be embedded within the 3′ untranslated region (UTR) of expressed reporter genes, facilitating their recovery with standard scRNA-seq protocols 29 , 46 , 47 , such that for each cell, historical information is recovered alongside an endpoint transcriptome. However, technical limitations of scRNA-seq, such as cellular loss, dropout and loss of spatial information, can introduce uncertainty or bias in the analysis of lineage as well as other recorded information. If scRNA-seq could enable recovery of all (or nearly all) recorded information from all (or nearly all) cells in the tissue or organism profiled, the inference of complete cellular histories would be possible. To our knowledge, the highest recovery that has been achieved while concurrently capturing DNA-based records was about 50% of cells in a monoclonal expansion of HEK293T cells in vitro 85 . An alternative to scRNA-seq would be for a rich set of recordings to be captured to a dense region and recovered by long-read sequencing, such that a single sequencing read would suffice for reading out the history of each cell.
Imaging-based methods can also recover omic measurements while fully preserving the spatial relationships of cells. Elegant approaches for querying thousands of genes use sequential rounds of fluorescent in situ hybridization and imaging 86 , 87 , 88 . Adaptations of such methods can be used to amplify and query single base edits in situ to recover information from DNA-based records 30 . Such methods could be adapted to discriminate between more diverse editing outcomes. For imaging-based spatial transcriptomics methods, this requires that the set of potential character states be known in advance, such that probes can be designed to discriminate between them. However, with sequencing-based spatial transcriptomic methods, diverse editing outcomes could potentially be read out directly 89 .
Reconstructing cellular histories
Lineage trees.
The development of tools to reconstruct cellular histories has been led by efforts to use data from constitutive recorders to generate cell lineage trees. Lineage tree reconstruction draws on the general principle that lineage relationships can be inferred by comparing edit patterns from constitutive recorders between cells (Fig. 2d ). Roughly speaking, the more similar the edit patterns are between cells, the more closely related the cells should be in the lineage tree. To enable accurate reconstruction, the properties of the recording technology have to be taken into account; for example, how character states are generated 90 , 91 , 92 and how the frequencies of different character states vary 9 , 90 , 91 , 93 .
Current algorithms for reconstructing lineage trees differ in speed and accuracy. Statistical methods, such as maximum likelihood or Bayesian methods, using an appropriate model can be very accurate. However, because the number of possible tree topologies grows super-exponentially with the number of cells, reconstruction of larger lineage trees that include many cells is typically not feasible because these methods are too slow. Instead, algorithms ensuring fast tree reconstruction using heuristics are used, although they may suffer from limitations in accuracy. For example, greedy algorithms, either building the tree top-down starting with early, shared indels 94 , or bottom-up by progressively merging cells with similar target sites 95 , can ensure reconstruction of large trees with the risk of finding only a locally optimal tree rather than the globally best tree. Alternatively, an iterative tree-building algorithm 96 first creates a basic tree structure using a subset of cells and then iteratively adds in additional samples. However, initial choices, especially those related to the tree’s backbone, may heavily bias the final tree structure. Systematic comparisons across tools with real and simulated benchmarking data are essential to evaluate the speed and accuracy of any construction algorithm 93 .
In addition to estimating an accurate tree topology, branch lengths can be calibrated to absolute time to understand the timing of various cell events such as differentiation or metastasis. Calibrating the lineage tree topologies in absolute time (such as hours) 90 , 95 can be achieved by modelling the edit accumulation as a function of time.
Cell population dynamics
Time-scaled lineage trees contain information about cell population dynamics. For example, if most branching events happened close to the start of the tree, the cell population probably experienced rapid early expansion and exponential growth. Alternatively, if most branching events occurred close to the end of the tree, then the cell population was probably fairly constant over time, which could be indicative of long-term maintenance of a stem cell pool 97 . The field of phylodynamics has built statistical tools to extract population dynamics from trees. In the context of single-cell biology, methods of estimating the population size and progenitor commitment times within the sampled population of cells 95 and the rates of cell division and death events in the entire cell population 90 have been proposed.
Intrinsic states, extrinsic signals and spatial context
Biological recording is possible and of interest even when it does not refer to cellular lineage information. Examples are signal event histories in a post-mitotic neuron (Box 2 ) or pathogen-exposed innate immune cell (Box 3 ). Ideally, the dynamics of multiple signals within a single cell’s history, in relation to one another as well as to absolute time and regardless of whether the cell is dividing, should be recordable and recoverable if ordered editing is assumed. However, presumably because multiplex biologically conditional editors are still very new, the development of algorithms for analysis of such data remains immature. There are a few datasets available, associated with the DOMINO, CAMERA and ENGRAM methods, that may serve as entry points or inspiration for computational approaches 64 , 65 , 78 . Potential framings of the problem and how it might be addressed algorithmically are discussed in the section ‘Practical challenges for DNA-based recording’.
Box 2 The potential of DNA-based recording in the nervous system
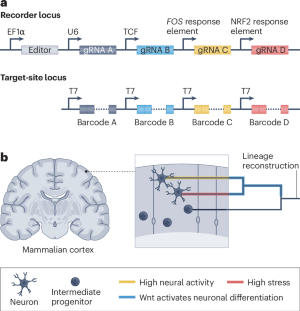
Understanding the complex processes that shape neural development and function is a central goal in neuroscience. By reconstructing the lineage history of individual neurons, we can gain insights into how the developmental relationship of a neuron with other neurons influences its final identity and function within the brain. Recording signalling pathway activities can further reveal the roles of specific pathways during cell-fate determination, while calcium activity and oxidative stress recordings can indicate how neurons respond to stimuli. For example, as illustrated, barcodes A, B, C and D could record lineage, Wnt signalling, calcium activity and oxidative stress, respectively (see the figure, panel a ). In such an experiment, constitutive editing of barcode A is first used to record the lineage relationships of neurons during mouse development, by expressing an editor from an EF1α promoter and a guide RNA (gRNA) under a U6 promoter. Conditional recording is then used to record the activity of Wnt signalling. A Wnt-responsive enhancer (TCF) is coupled to the activation of an orthogonal editor that introduces edits into barcode B. Next, conditional recording is used to record neural activity in postmitotic neurons. Long-term calcium activity is recorded by coupling the immediate early gene FOS to another orthogonal editor that introduces edits into barcode C. Conditional recording is then used to record oxidative stress. An NRF2 (nuclear factor erythroid 2-related factor 2)-binding DNA response element, which responds to electrophiles and reactive oxygen species, is coupled to the activation of another orthogonal editor that introduces edits into barcode D. The behaviour of the mouse would also be recorded for the duration of the experiment. Finally, individual neurons are isolated and barcodes are transcribed by T7 RNA polymerase and then sequenced together with single-cell transcriptomes. Integration of data from the four barcodes and the cellular identity is used to reconstruct the history of each neuron (see the figure, panel b ). Notably, the individual components of the proposed recorders already exist as reporter genes but would be combined here to record information into DNA.
Box 3 The potential of DNA-based recording in the immune system
Innate immune cells can remember past encounters with pathogens, enhancing their responsiveness upon re-exposure 200 , 201 . This phenomenon, termed trained immunity, expands our understanding of innate immune function and its importance in host defence. However, many questions remain regarding the specific immune-cell populations involved, the metabolic changes that occur, such as glycolysis, and the duration of the memory response. These questions could be addressed by a DNA recording system with four barcodes (BCs) at target sites specified by guide RNAs (gRNAs), as illustrated (see the figure, panel a ). In this example, a DNA recorder constitutively edits barcode A to capture lineage information of the innate immune cells during their specification and differentiation. Another editor with a transcription factor coupled to pattern recognition receptors (PRRs), which detect pathogen-associated molecular patterns (PAMPs), translocates into the nucleus upon activation, where it regulates editing of barcode B 202 , 203 . Glycolysis, a key metabolic pathway upregulated in trained innate immunity, is monitored with a glycolytic biosensor detecting metabolites such as fructose-1,6-bisphosphate, initiating conditional editing of barcode C 204 , 205 , 206 , 207 . In addition, the pro-inflammatory signalling response would be recorded in barcode D under the control of NF-κB response elements 208 , 209 . Innate immune cells could then be sequenced to recover their cellular identities and recorded histories, providing a comprehensive view of the trained immunity process (see the figure, panel b ). The reconstructed information would allow researchers to infer the number of pathogen challenges the cells encountered, the metabolic and pro-inflammatory response of the cells, and how these responses differ across cell types. This information would in turn provide insights into the formation of immune memory and the responses of trained innate immune cells upon pathogen re-exposure.

Tree decoration
Combining the cell lineage tree with the cell’s past trajectory, its extrinsic signals and its spatial context information into a cellular history involves decorating the tree with this information obtained from conditional recorders as well as endpoint measurements. Provided that the constitutive and conditional recording occurs concurrently, the tree would provide guidance on when the signals occurred. For example, if two sister cells (relationship informed by constitutive recording) share a recorded signal (obtained from conditional recording) but no other cells do, then the event recorded may have occurred in the parent of the two sister cells. Endpoint measurements can enrich such conditional recorder data by providing the state of each cell at the end of the experiment.
Rich datasets in which cell lineage, trajectory information, extrinsic signals and spatial context are concurrently recorded are not yet available. At present, decorating a tree relies merely on molecular state information of the analysed cells at the end of the experiment. Two primary methods have thus far been developed for such data: first, methods estimating transition maps between cell states from several time points, incorporating some (but by far not all) lineage information 98 , 99 , 100 ; and second, methods that decorate the tree at ancestral nodes. For example, a maximum-parsimony approach has been applied to infer tissue location within tumour phylogenies 8 .
Accounting for noise and uncertainty
Phylogenetic reconstruction and tree decoration methods include heuristic, distance-based, parsimony-based methods 18 , 29 , 47 , 94 , 96 , as well as statistical maximum-likelihood and Bayesian methods 90 , 95 . Major challenges for reconstruction stem from the noise in the data, owing to heterogeneities in reading and writing as well as incompleteness of the recording data. Noisiness for a given cell may stem from silencing or imperfect readout, leading to ‘drop out’ of recorded information at some sites. Furthermore, recordings for only a subset of cells are likely to be captured and not every division of cells may be recorded (that is, sampling depth is limited). In general, methods can deal with incompleteness by imputing missing data 94 , 95 , by explicitly modelling the loss of information 90 , or by subsampling a set of cells from the tissue 90 . The uncertainty resulting from heterogeneous and incomplete data can be considered by Bayesian phylogenetic approaches or through phylogenetic bootstrapping to obtain confidence estimates.
Heuristic methods 96 are typically fast, enabling estimation of lineage trees on millions of tips. However, the estimates may be rather uncertain or biased owing to the noise in the data and heuristics may fail to converge to global optima. By contrast, statistical methods using an appropriate model can lead to unbiased results with quantification of uncertainty. In particular, Bayesian methods naturally incorporate noise and uncertainty, but are very computationally intensive, only facilitating analysis of a few hundred cells to a couple of thousand cells. As recording becomes complex, the noise and uncertainty in the data will decrease, and non-statistical frameworks may lead to reliable decorated trees. We anticipate that expansion of recording capacity will alleviate some statistical and computational challenges.
Applications of DNA-based recording
The application of DNA-based recording to generate biological insights remains in its early stages. Our selected examples illustrate the use of DNA-based recording to investigate: 1) the lineage origins of various cell types; 2) the dynamics of clonal dominance in development and disease; 3) the orchestration of cell-fate decisions by intrinsic and extrinsic signals; and 4) non-invasive bacterial monitoring of cellular behaviours and environmental conditions (Fig. 3 ).

a , CRISPR-based DNA recording traced the origins of regenerated cardiomyocytes following heart injury in zebrafish. Single-cell RNA sequencing (scRNA-seq) at defined days post-injury (dpi) retrieved DNA recordings concurrent with cell states. These data enabled the reconstruction of lineage relationships, identification of cell types, and point to the epicardium as a likely source of col12a1a -expressing fibroblasts. b , CRISPR-based DNA recording reveals clonal relationships in cancer progression through a xenograft study involving human KRAS -mutant lung adenocarcinoma cells transplanted into the left lung of immunodeficient mice. Analysis of dissected and sequenced metastatic cells identifies key genes contributing to metastasis by integrating clonal relationships with spatial cell-state information. c , Conceptual diagram of a recombinase-based circuit reporting morphogen activity during gastruloid formation. A morphogen-responsive enhancer drives the expression of rtTA (reverse tetracycline-controlled transactivator). In the presence of doxycycline (dox), rtTA activates a Tet-responsive element, driving expression of Cre recombinase. Cre-mediated recombination at loxP sites removes a stop cassette enabling constitutive expression of a fluorescent reporter. This approach allows the visualization of cells that have experienced morphogen signalling within a defined recording window, revealing spatial patterns of signalling pathways coinciding with gastruloid symmetry breaking and elongation. d , Sentinel Escherichia coli engineered with Record-seq reverse-transcribe transient mRNA into CRISPR arrays within the gut using Cas1–Cas2-based recording. Analysing faecal samples through deep sequencing and computational methods offers insights into bacterial adaptation to gut conditions such as dietary shifts and inflammation response. CRISPR, clustered regularly interspaced short palindrome repeats; RT, reverse transcription. Part a adapted from ref. 7 , CC BY 4.0 ( https://creativecommons.org/licenses/by/4.0/ ). Part b adapted with permission from ref. 8 , AAAS. Part d adapted with permission from ref. 12 , AAAS.
Lineage origins during development and regeneration
A central goal of developmental biology is to elucidate cell lineage relationships and molecular changes during the development of a single-celled zygote into a multicellular organism composed of numerous cell types. Most studies to date have focused on clonal tracing, which defines the descendants of a cell 5 , 6 , 7 , 101 , 102 . For example, a major question in blood development and immunology is what kinds of progenitor give rise to diverse blood and immune cell types and how their abundance changes with age. To address this question, an inducible scarring-based barcoding system, CARLIN, was used to label mouse cells in vivo with a unique genetic barcode, which was then captured alongside the transcriptome via scRNA-seq 5 . Prior to this work, the general paradigm was that definitive haematopoietic stem cells (HSCs) derive from pre-HSCs and establish blood production in late fetal and adult mice. However, lineage tracing facilitated the identification of a new progenitor type, embryonic multipotent progenitors (eMPPs), which are also derived from pre-HSCs. Barcoding analysis revealed that eMPPs preferentially contribute to lymphoid lineages and persist lifelong, but that their output decreases with age. Conversely, adult HSCs increase productivity with age but do not compensate for the loss of lymphoid cell production from eMPPs. These data, obtained by recording, provide a potential explanation for immune decline during ageing.
Another scarring-based method, called LINNEAUS, was used to investigate the origin and functions of key cell states in zebrafish heart regeneration 7 . A key question in regeneration is which cells contribute to the regenerated tissue. Three transient fibroblast cell types induced by heart injury were identified to have pivotal roles in tissue repair. DNA-based recording revealed that two types of fibroblast (expressing col11a1a or col12a1a ) were lineage-related and originated from the heart epicardium, whereas the third type (expressing nppc ) emerged from the endocardium (Fig. 3a ). Depleting col12a1a -expressing fibroblasts impeded heart regeneration, underscoring their pro-regenerative function. The study showcased how DNA recording helps to reveal the distinct origin and signatures of cells with regenerative functions.
Clonal dominance in development and cancer
Embryonic progenitors can give rise to clones of drastically different sizes, thanks to a combination of intrinsic differences, extrinsic cues and stochastic processes 103 , 104 . Recording approaches as well as live imaging have revealed clonal dominance, where a small number of progenitors contribute disproportionately to a specific organ or cell type. For example, the DNA scarring-based method GESTALT was used to barcode embryonic progenitors to trace lineage throughout zebrafish development 9 . Remarkably, while about 20 early embryo cells seemed to give rise to the entire zebrafish blood system, as few as 5 clones explained 98% of the blood lineage in an adult animal. This study illustrates the power of DNA-based recording to reveal patterns of clonality from embryo to adult.
Clonal dominance is also evident in cancer, where evolving clones within tumours acquire specific survival, proliferation and metastatic properties. Classic studies showed intratumoural heterogeneity between primary and distant sites but did not reveal how cancer metastasis plays out within individual clones over time. Lineage tracing using DNA recorders has been recently applied to yield insight into clonal evolution and the role of specific genes in metastasis and cancer progression 8 , 105 , 106 , 107 , 108 . For example, human lung cancer cells, engineered to record their lineage by DNA scarring, were surgically implanted into the lungs of mice 8 . Phylogenetic tree reconstruction was performed for hundreds of clones as they grew and spread over months using the mutated barcodes. Reconstructing deep trees and analysing where related cells were positioned within the lung allowed quantification of metastatic capacity for each clone , which ranged from completely non-metastatic to the aggressive colonization of distant sites (Fig. 3b ). This analysis also revealed a metastatic hub in lymph tissue, and complex seeding topologies disseminated clones across the body and even back to primary tumours. Gene-expression differences between clones arose early before transplantation and were mildly predictive of these divergent metastasis phenotypes, except for rare exceptions where some clones developed new potential. Candidate gene perturbations also altered invasiveness phenotypes, validating their functional role in driving metastasis. Overall, this work, as well as several other related studies 105 , 106 , 107 , 108 , demonstrates the power of DNA recorders to illuminate metastatic progression and heterogeneity.
Intrinsic and extrinsic cellular experiences
A key challenge in developmental biology is to disentangle the effects of cell-intrinsic factors inherited through lineage and cell-extrinsic signals in the local cellular environment on cell-fate decisions. The ability to reconstruct lineage in situ could reveal the relative roles of the two kinds of cue. To this end, the image-readable, recombinase-based recording tool intMEMOIR was used to trace the emergence of fly neuron types from an embryonic progenitor pool 10 . Imaging-recombined barcodes in adult brains revealed that neurons derived from the same embryonic clone exhibited a spatially dependent similarity in terminal cell identity, with nearby cells more similar in cell type than those located further apart. By contrast, cells from different clones showed no relationship between their spatial position and terminal cell identity. These findings highlight the complex way in which lineage and extrinsic cues combine to determine cell fates in a spatially patterned tissue.
Another fundamental question in developmental biology is how symmetries are broken and axes are formed in a stereotyped fashion. Stem-cell-derived embryo models provide experimentally accessible systems to address this question. A recent study used DNA rearrangement-based recorders in the mouse gastruloid model to conditionally record signalling activated by Nodal, Bmp or Wnt morphogens 11 . The DNA rearrangements produced heritable changes in fluorescent protein expression, enabling live imaging of recorded signals during gastruloid development. This approach revealed that Nodal and Bmp exhibited spontaneous heterogeneous activity among cells, which modulated Wnt signalling. Differences in Wnt signalling led to the emergence of distinct Wnt-active and Wnt-inactive cell populations, which differed in their expression of the adhesion molecules that allow sorting into posterior and anterior domains (Fig. 3c ). Interestingly, recording at different times revealed that Wnt activity was only weakly predictive of the later spatial position along the anterior–posterior axis when recorded at 72 hours after gastruloid formation, but became strongly predictive when recorded at 96 or 120 hours after gastruloid formation. These results demonstrate that Wnt activity is a critical player in symmetry breaking, and acts as a predictive factor for future cell positions and fates along the anterior–posterior axis.
Non-invasive recording of the environment
The examples discussed above are focused on capturing lineage relationships among mammalian cells, except for the example in ref. 11 , which reports recording or marking of canonical signalling pathways. Another example illustrates recording of an additional non-lineage aspect of cell states, in prokaryotes rather than eukaryotes. Specifically, bacteria were recently engineered to record their own global transcriptional responses during transit through the mouse gut 12 . A chimeric Cas1–Cas2-RT (reverse transcriptase) insertional editor was used to reverse-transcribe cellular mRNA and integrate the resulting DNA into a genomic target site. These engineered bacteria were deployed as ‘sentinels’, as the recorded transcripts were informative with respect to their interactions with food, host cells and other microbes during gut transit. The recordings were recovered through RNA-seq of samples collected from faeces and revealed bacterial adaptation to nutrient availability, acid stress, inflammation and microorganism–microorganism interactions (Fig. 3d ). Although currently limited to Escherichia coli , this study and other Cas1–Cas2-based recording systems 81 open up possibilities for non-invasive analysis of complex microbiota physiology and adaptation in vivo.
Practical challenges for DNA-based recording
These initial demonstrations of recording serve as motivating prospects for what could be achievable, and eventually routine, as we develop more powerful recording systems. However, to realize their full potential, the field must address several key technical challenges to writing, reading and reconstructing DNA-based recordings.
Challenges for writing information
Programming conditional recorders.
To date, most work in this field has been based on constitutive recorders. Ideally, these would be complemented by a library of conditional recorders that respond to signalling pathway activity, enhancer activity, specific transcripts, intracellular ligands, extracellular ligands, metabolic fluxes, neural activity, mechanical forces and infections, among other biological parameters. In practice, only a few studies have demonstrated conditional recorders, mostly relying on signal-dependent cis -regulatory elements (CREs) to drive production of a gRNA or pegRNA, or transcriptional acquisition systems in prokaryotes 11 , 12 , 19 , 78 , 80 , 81 . Much work remains to be done to expand and optimize the repertoire of CRE-based recorders, for example, to encompass all major signalling pathways and cell types, while transcriptional acquisition systems beg to be adapted to eukaryotic systems. Furthermore, existing signal-dependent CREs may not report linearly on pathway activation, and concatemerized synthetic reporters might behave differently from endogenous response elements. Furthermore, many biological phenomena occur post-transcriptionally, requiring alternative strategies to activate conditional recorders. For example, dual-component gRNAs can be used to create conditional recorders activated by specific protein–protein interactions 79 . Leveraging de novo designed binders or receptors, or aptamer-driven gRNA activation, could further expand the range of recordable signals. Finally, we note that these early demonstrations of conditional recorders have yet to be coupled to constitutive recorders.
Maintaining bio-orthogonality
Editors, recorders and recording media must maintain bio-orthogonality, that is, they must minimize interference with native biology. Although most cells and organisms can tolerate the extensive addition of genomic content (for example, 34 megabases (Mb) in the TcMAC21 mouse model, a transchromosomic mouse model of Down syndrome that harbours the long arm of human chromosome 21 as a mouse artificial chromosome (MAC) 109 ), suggesting the potential for accommodating substantial amounts of recording ‘equipment’, each biological model may interact differently with any recording components used. The interference can result in low portability (for example, low efficiency of Cas1–Cas2-based editing in mammalian cells), toxicity and off-target effects (for example, Cas9-induced DSBs 50 , 53 or the Cre recombinase 110 ), or disruption of genomic context around the integration sites of recording components. Although disruption of the genomic context can be mitigated using defined ‘safe harbour’ loci to integrate editors, recorders or media, addressing the former two issues requires thorough testing and validation using the biological model of interest. In the future, de novo designed binder or receptor proteins could potentially be used as sensors and/or signal transduction systems to help to maintain bio-orthogonality 111 .
Timescales and capacity
Cellular histories with single-cell-cycle resolution require at least one edit per cell division. A key challenge is stabilizing the edit rate, which can diminish over time owing to factors such as silencing of editor expression and reductions in the number of unedited target sites. Ideally, editors and recorders would reside at safe harbour loci to resist silencing, and the system would possess vast amounts of information capacity or use mechanisms to stabilize the recording rate (for example, with DNA Typewriter, each successive write event both destroys and creates an editable site 76 ). Recorders should ideally vary across timescales from capturing sub-cell-cycle events in seconds or minutes (for example, cell-signalling cascades) to monitoring long-term processes over hours, days, months or years (for example, cellular differentiation or disease progression). Since DNA-based recorders rely on DNA repair mechanisms, they are inherently more compatible with the longer timescales, whereas recorders based on other modalities such as RNA or proteins may be better suited to fast timescales.
Spatial information
Current spatial methods capture only a single moment in cellular interactions, lacking insight into the spatial origins of cells. In organs such as the brain and immune system, understanding past spatial proximity is crucial for subsequent development and function. For example, recording spatial proximity could shed light on migration patterns during embryogenesis or reveal anatomical locations and previous cell contact histories of immune cells 112 , 113 . It could also allow the spatial inference of metastasis from initial tumour to colonization of new tissues. One possible way to record the spatial trajectory of cells is to leverage synthetic signalling pathways. In this approach, ‘sender’ cells expressing synthetic ligands activate matching synthetic receptors in neighbouring ‘receiver’ cells, for example, SynNotch, Tango, MESA and BAcTrace 114 , 115 , 116 , 117 , 118 , 119 , 120 , 121 , 122 , 123 . SynNotch is a promising approach because it labels neighbouring cells upon contact and could potentially be coupled to CRISPR recorders. However, a challenge lies in ensuring unique labelling of each receiver cell by each sender, requiring numerous orthogonal signalling channels or intercellular genetic material transfer.
Recording across model and non-model organisms
Understanding the evolution of development and what features are conserved or variable between species remains one of the most fundamental challenges in biology. Generating stable transgenic cells and animals with diverse recording capabilities will allow rapid characterization and comparisons of development and homeostasis across various organisms. However, complex, stably integrated recording systems may be difficult to engineer in most non-model organisms. Creating ‘portable’ recording systems that can be introduced into diverse animal species using viral vectors could address this limitation. These systems need to be genetically compact yet provide enough memory to record sufficient data to analyse particular developmental processes, and to maintain bio-orthogonality with respect to the native biological processes. The development of larger and more tissue-specific delivery vectors 124 , 125 , 126 , as well as artificial chromosomes with large capacity, high mitotic stability and broad host range should help to realize this possibility 127 , 128 , 129 .
Challenges for reading DNA records
Missing information.
Many of the outstanding technical challenges for recovering DNA-recorded information are touched on above (see section ‘Reading DNA-recorded information’), and essentially converge on missing information, that is, a failure to recover all information recorded in each cell (drop out), or to recover histories of all cells from a tissue or organism (sampling depth). The consequences of missing information can be substantial. For example, recovering just 1% of cells from an organism allows the generation of sparse trees that are informative of early lineage relationships, but it is uninformative for the key terminal cell divisions that drive cellular specialization. Taking the adult mouse as an example, recovering DNA records with almost 100% efficiency from nearly all of its approximately 10 billion cells is a task on a scale that cannot currently be achieved, even if the sampling depth and dropout issues were fully solved, owing to limitations inherent in scRNA-seq technology. Therefore, the field needs scRNA-seq methods to advance by several orders of magnitude, spatial transcriptomic methods to advance to a level at which entire animals can be routinely profiled at single-cell resolution, or DNA-based recording to advance to the point where the entire, longitudinal history of a cell can be captured by a single, contiguous DNA sequencing read.
Survivorship bias
Another key challenge related to recovering DNA records is survivorship bias — many if not most cells produced during an organism’s life die, resulting in the loss of their records. Moreover, current approaches rely on isolating and destroying cells that contain recorders. One way these issues could be addressed is by engineering cells to export recordings over time through protective nanoparticles. For example, the COURIER system uses RNA export systems based on viral and synthetic components that efficiently package target RNAs into protective nanoparticles secreted from cells 130 . By incorporating RNA barcodes, sampling the exported RNA from culture media, and sequencing these barcodes enables longitudinal tracking of clonal population dynamics and overcomes barriers to accessing RNA from living cells in a non-destructive manner. Furthermore, if each cell produced multiple nanoparticles before division, this amplification could reduce the likelihood of cell dropouts. This approach resolved the expansion and decline of thousands of distinct cell clones over time in response to drug selection. These export tools have versatile applications, including real-time monitoring of biological phenomena for early disease detection and treatment optimization. In the future, increasing the rate at which nanoparticles are generated would help to resolve biological processes that occur at faster timescales.
Challenges for reconstructing histories
Quantifying cell dynamics.
An overarching challenge is to accurately quantify variations in cell behaviour and thus to capture cellular dynamics, such as the rates of division, differentiation, apoptosis and migration. Although similar cells tend to exhibit similar dynamics, certain factors such as gene-expression variability among cells or responses to signalling molecules can have a more pronounced influence on cell dynamics than others. The phylodynamic framework should enable such quantification based on a reconstructed decorated tree 131 . As phylodynamic approaches often require in-depth adjustment even within their field of origin — epidemiology — reasonable model assumptions and approximations remain to be explored for biological recording data.
Computational scaling
The computational cost of assembling trees grows exponentially with the number of cells, necessitating more scalable computational approaches to phylodynamic inference. Some approaches developed recently for datasets generated during the COVID-19 pandemic, which contain millions of sequences, may become helpful 132 , 133 , 134 . First, one can use fast heuristics for tree topology estimation and, if appropriate, ignore all topology uncertainty, while using statistical tools that provide an estimation of uncertainty for more uncertain processes like timescales. Second, for phylodynamic parameter estimation, analyses with smaller numbers of cells might be informative, and analyses of smaller subtrees could be merged for overall results. Third, utilizing graphics processing units (GPUs) for calculations and large memory can further facilitate analyses.
Timings of conditional recordings
A conditional recorder captures specific features of a cell’s history over time, with the intensity of recording dependent on the abundance of that feature (for example, RNA transcript or signal transduction activity levels). To interpret such data accurately, we need to establish the timing of conditional recordings, otherwise we cannot distinguish between short periods of high signal intensity versus long periods of low signal intensity, nor between the relative timing of different signals in the same system if multiple conditional recorders are present. One strategy to address this issue is to implement a constitutive time recording that runs in parallel to the conditional recording 17 . Lineage tracing through a constitutive recorder would allow conditionally recorded events to be mapped back on a branch in the lineage tree, albeit with a temporal resolution limited by the rate of cell division. However, with recording strategies that explicitly preserve the order of events in writing to DNA and furthermore write constitutive and conditional signals to a shared medium 76 , 78 , duration versus intensity versus order of conditional signal(s) could be disentangled even between cell divisions or in post-mitotic cells.
When analysing such data, it will be necessary to consider whether a given set of cells or recordings share a common history. For sets of cells or recordings without a shared history, such as non-dividing neurons, time-series analysis approaches might be applicable 135 , 136 . If the cells or recordings share a common history, it will be important to account for this shared lineage to avoid biases 137 . Ideally, one would perform joint analysis of cell lineage and conditionally recorded signals, given that they may be mutually informative. Packer et al. 138 leverage Caenorhabditis elegans to provide a compelling early example of how this might be approached, by jointly analysing single-cell transcriptome data layered onto the invariant lineage. For such joint analyses, approaches developed for time-lapse microscopy data can also be considered 139 . However, additional complexities arise with DNA-based recording data. These include the need to reconstruct rather than directly image the cell lineage tree, the indirect measurement of the timing of features through a separate recorder, and the fact that feature intensity is inferred from the number of recordings within a given time window.
Combining datasets
Complete molecular recording over vast spatiotemporal scales is currently not feasible. However, integrating datasets from different experiments, using data from the same biological entity but different replicates, time periods and/or molecular modalities has the potential to yield a cohesive view of the cellular dynamics governing a system of interest. Combining data from different individuals of the same species may allow us to differentiate deterministic rules from stochastic fluctuations and identify ‘historical’ molecular and cellular events underlying phenotypic changes due to mutations. It could also facilitate comparative developmental analyses across species, identifying both conserved and species-specific developmental programs. However, the development of methods for combining data across individuals or experiments in a coherent way remains a substantial challenge.
Infrastructure and standards
Accessible computational tools will be essential for advancing these developmental recording techniques, akin to the impact of ready-to-use computational analysis platforms for scRNA-seq or pathogen data analysis. As recording systems proliferate, and data accumulate, it will also be essential to develop public repositories and data standards, analogous to successful platforms in other areas of biology such as NextStrain ( https://nextstrain.org/ ). These resources will enable data sharing, visualization and exploration of DNA-based recording datasets across methods and model organisms.
Emerging opportunities
In this section, we ask what exciting possibilities lie ahead. We focus on four domains: (1) high-capacity recording of development and homeostasis; (2) understanding the statistical nature of developmental programmes; (3) causal inference within and across individuals; and (4) engineering recorders to provide sentinel cells in humans.
Dense recording of development and homeostasis
We have yet to come even remotely close to saturating the enormous theoretical capacity for DNA-based information storage in living cells and organisms while maintaining viability. As noted above, mouse models with artificial chromosomes as large as 34 Mb are viable 109 , and the upper limit on how much engineered content can be added while maintaining viability has not been seriously explored. As a thought experiment, imagine that about 1% of the mouse genome (25 Mb) were engineered to support the data-storage aspect of biological recording. At a modest density of 2 recorded bits (that is, one base) per 50 base pairs (bp), this configuration would provide about one megabit of storage per cell. Although actual developmental lineage trees tend to be asymmetric, for simplicity, if we assume a perfect binary tree of 40 cell cycles from fertilized zygote to the 10 10 cells or so that constitute an adult mouse, only a small fraction of this storage capacity is sufficient to completely capture cell lineage relationships. For example, if ordered recording was enabled, and 8 bits were successively set at random at each cell division to distinguish daughter cells from one another (2 8 = 256 possibilities), only 320 bits would be required to capture a complete lineage tree from zygote to adult. In this scenario, over 99.9% of that one megabit would remain available to record aspects of biology other than cell lineage via conditional recorders (Fig. 4a ).

a , A mouse model with around 1% of its genome devoted to encoding recorders that respond to input signals and another 1% or so dedicated for sites targeted by their corresponding recorders would enable as many as 2,500 signals to be concurrently recorded in single cells. b , Stereotypical structures such as the mammalian retina consist of a conserved set of cell types in defined ratios, yet how this is reproducibly established during development remains unclear. Cell histories recovered through DNA recordings would help to reveal the mechanisms by which heterogeneous clones give rise to uniform structures, how these vary between homologous structures within the same individual (such as the left and right eye), and how these vary between individuals. Analysing variability at multiple scales should reveal the statistical rules that operate to generate stereotypical fate distributions and tissues. gRNA, guide RNA.
If an additional 1% (25 Mb) or so were devoted to encoding the recorders themselves (not the target sites), at a modest density of one recorder per 10 kilobases (kb) or so, as many as 2,500 recorders could operate concurrently within each cell. A handful would be constitutive recorders, but the remainder could be conditional on signalling pathways, cell-type markers, epigenetic states, enhancer activities, transcript levels, intracellular and extracellular ligands, metabolic fluxes, neural activity, mechanical forces, infections or other aspects of cell biology. As such, it is possible to imagine a vast array of time-resolved internal recordings recoverable from individual cells. These systems could also be used to record information passing between cells. For example, systems could record cell–cell interactions or neuronal connections. The resulting datasets would realize the vision of a densely decorated lineage tree with reconstructed signal dynamics (Fig. 1c ). Furthermore, ordered recording could enable the capture of quantitative dynamics in the absence of cell divisions, for example, in post-mitotic neurons or adult homeostasis, over weeks, months or years. Overall, assuming we exhaust our hypothetical DNA-based storage capacity over the course of the lifetime of an adult mouse composed of around 10 10 cells, we could in principle capture as much as 2 53 bits, or more than a petabyte, of information, per individual.
Statistical development
Variable development is the rule; the famous invariance of C. elegans lineage is an exception. The robustness with which development unfolds in most multicellular organisms is a consequence of stochastic rather than deterministic processes. However, this has been deeply explored in only a handful of cases (for example, branching morphogenesis 140 ). In most contexts, the statistical analysis of development is substantially constrained by our ability to track the state of individual cells at scale. For example, in the vertebrate retina, individual progenitor cells give rise to variable clones, both in terms of size and cell-type composition. How these variable clones together create the stereotypical and reproducible structure of the retina is still an open question. Patterns in the distribution of cell fates on lineage trees can reveal the mixture of fate-restricted progenitors that in turn generate the full distribution of cell fates in the mature retina with the right proportions and spatial organization 141 . Zooming out, reconstruction of decorated lineage trees may provide fundamentally new ways of addressing some of the oldest questions in developmental biology, for example, the statistical and signal-enforced rules by which fate-biased progenitors collectively generate the cell-type distributions required for functional modules (for example, a nephron or neuronal circuit), tissues (for example, a retina) or organs (for example, a liver) (Fig. 4b ). Even within a single individual, comprehensive recordings may enable us to distinguish the stochastic versus constrained aspects of such processes (for example, how many ways there are to make a nephron).
Of course, variation will manifest not only within a single individual but also between individuals, and both within genotypes as well as between phenotypes. To what extent can phenomena such as incomplete penetrance and variable expressivity be explained by the variance induced through the statistical rules underlying development? To what extent can common phenotypic variance among humans be understood as a consequence of the same statistical processes? Through the analysis of dense, decorated trees generated from many individuals, both within and across genotypes, it may be possible to extract general principles about how macroscopic phenotypic variance emerges from earlier stochastic events in the context of normal development 142 , 143 .
Causal inference
Although we understand the genetic basis for thousands of Mendelian disorders, our understanding of the mechanisms by which mutations in particular genes give rise to specific phenotypes lags far behind. Developmental disorders are mostly pleiotropic; a multitude of system- and context-specific mechanisms can underlie the path from genotype to phenotype. Historically, the biology of developmental disorders has been studied at a limited number of timepoints, typically after a phenotype has manifested. This approach offers little insight into the early moments of causation that are buried within the developing embryo. For many disorders, combining densely decorated lineage trees from different timepoints and comparing mutant and wild-type trees could potentially pinpoint the molecular and cellular causes of downstream phenotypes (Fig. 5a ). For example, shifts in statistical rules governing particular cell types at particular moments might explain how the haploinsufficiency of a gene leads to each pleiotropic aspect of a gross phenotype.

a , Leveraging DNA recorders to elucidate how changes in cell-type proportions arise in diseased states. Here, a synthetic circuit allows recording of transcription factor (TF) A over time and reveals how the late onset of TF A in diseased cells results in aberrant cell-type proportions. Synthetic circuits could also be used in combination with perturbation screens, in which various genes could be perturbed at different time points and/or cell types to understand how specific signalling dynamics result in particular phenotypes. b , Sentinel cells engineered with DNA recorders are introduced into patients, where they localize to different organs or tissues and systematically capture diverse molecular, cellular and physiological cues within their spatial environment. These sentinel cells can be programmed to trigger responses upon detection of specific signals to eliminate pathological cells that may evade the immune system. They can further be programmed to halt their responses upon detection of normal homeostasis in recovered, healthy tissues. Subsequent sequencing (seq) of sampled sentinel cells would unveil a comprehensive disease history.
In the future, organism-scale biological recording systems could potentially be combined with synthetic biology approaches to activate user-defined genetic circuits at specific times or within specific sublineages. During development, synthetic circuits could be used to track cell states and ectopically activate transcription factors to drive desired downstream fates or morphological characteristics. This paradigm could be used to generate novel cell types with hybrid functions: for example, pancreatic alpha or delta cells that, in addition to their normal hormone outputs, secrete insulin, or retinal cells with rod-like sensitivity but with cone-like colour detection. Alternatively, in physiology, neurons could be selectively activated according to their prior activity levels stored through recordings. This would enable analyses of how memory and learning are established in the brain, and reveal to what extent prior activity has an impact on future responses. We can also imagine future cellular recording systems that are combined with multiplex in vivo perturbation systems to perturb multiple genes in multiple timeframes in multiple cell types, all within the context of a single animal, followed by recovery and analysis of the molecular and cellular consequences for each of these perturbations of gene, time frame or cell type.
Engineered memory sentinel cells in humans
Recording molecular and physiological states over time in individual people would provide rich datasets that enable early diagnosis, improved treatment of disease and better-informed health decisions, as well as insight into mechanisms and causes of disease. Optical and electronic devices are increasingly being deployed for non-invasive monitoring of blood glucose and some other basic physiological variables. However, most signals in the body, and the variations in those signals across tissues and organs, are inaccessible with such devices. Assuming that safety concerns could be adequately addressed, an alternative paradigm would be to engineer autologous ‘memory sentinel cells’ that reside within the body at different locations, and passively record a diverse range of molecular and cellular biomarkers (Fig. 5b ). These cells could be engineered to allow readout of the signals through reporter systems consisting of secreted information-dense protein or nucleic acids retrieved from urine, blood or stool 130 , 144 . Memory sentinel cells would thus provide the possibility of surveilling all organ systems, analogous to a human-readable immune system. For example, in slow, degenerative disorders such as Huntington disease or age-related macular degeneration, sentinel cells could store and report longitudinal information, such as extracellular biomarkers and other microenvironmental parameters, alongside tissue-location information, to provide insights into the spatiotemporal mechanisms behind disease progression.
Additionally, memory sentinel cells could be extended to analyse and act upon the information they record (Fig. 5b ). Such active memory sentinel cells could perform complex logical operations and enact interventions more precisely than what is possible with current approaches, such as chimeric antigen receptor (CAR) T cells. They could also potentially incorporate mechanisms to address safety concerns, for example, by incorporating engineered tumour suppressors or small-molecule-activated self-destruct circuits to serve as ‘kill switches’. Recording could provide two inter-related advantages for such cells. First, it would enable the cell to store large amounts of sensing data in the genome and access those data to make decisions about whether and how to respond in a given context. This could enable targeting of pathological microenvironments defined by complex combinations of factors, ensuring that treatments are delivered only where needed. Second, by recording data over long timescales, a cell could respond not only to the present state of their environment but also to its dynamic history, identifying aspects of the physiological state that are worsening or improving. If the sentinel cells migrate or circulate, this dynamic history could integrate information over space and time. This would allow engineered cells to carry out specific functions in one tissue informed by information recorded in other tissues or organs. More generally, such cells, with complex programmable logic and access to a large memory, could execute programs whose complexity vastly exceeds what is currently envisioned in synthetic biology and cell therapy fields.
Conclusions
Here, we have considered the rationale for biological recording, the state of the art of the underlying technologies, applications of this paradigm so far, various practical challenges that are presently rate-limiting and emerging opportunities. We envision that recent technological advances in DNA-based recording, coupled with overcoming these challenges, will enable the routine recording of cellular histories. This approach will further our understanding of cell decisions over time, given their cellular ancestry and past cell trajectories, and in response to external signals and spatial context. Comparative analyses of such data will enable the deduction of general rules of cell decisions in development and disease. Looking ahead, when recordings can be coupled to responses, such technologies will offer unique opportunities within synthetic biology, in both basic research and in translation to medical applications.
Woodworth, M. B., Girskis, K. M. & Walsh, C. A. Building a lineage from single cells: genetic techniques for cell lineage tracking. Nat. Rev. Genet. 18 , 230–244 (2017).
Article PubMed PubMed Central CAS Google Scholar
Zon, L. I. Intrinsic and extrinsic control of haematopoietic stem-cell self-renewal. Nature 453 , 306–313 (2008).
Article PubMed CAS Google Scholar
Livesey, F. J. & Cepko, C. L. Vertebrate neural cell-fate determination: lessons from the retina. Nat. Rev. Neurosci. 2 , 109–118 (2001).
Schürch, C. M. et al. Coordinated cellular neighborhoods orchestrate antitumoral immunity at the colorectal cancer invasive front. Cell 182 , 1341–1359.e19 (2020).
Article PubMed PubMed Central Google Scholar
Patel, S. H. et al. Lifelong multilineage contribution by embryonic-born blood progenitors. Nature 606 , 747–753 (2022).
Nusser, A. et al. Developmental dynamics of two bipotent thymic epithelial progenitor types. Nature 606 , 165–171 (2022).
Hu, B. et al. Origin and function of activated fibroblast states during zebrafish heart regeneration. Nat. Genet. 54 , 1227–1237 (2022).
Quinn, J. J. et al. Single-cell lineages reveal the rates, routes, and drivers of metastasis in cancer xenografts. Science 371 , eabc1944 (2021).
McKenna, A. et al. Whole-organism lineage tracing by combinatorial and cumulative genome editing. Science 353 , aaf7907 (2016).
Chow, K.-H. K. et al. Imaging cell lineage with a synthetic digital recording system. Science 372 , eabb3099 (2021).
McNamara, H. M., Solley, S. C., Adamson, B., Chan, M. M. & Toettcher, J. E. Recording morphogen signals reveals origins of gastruloid symmetry breaking. Nat. Cell Biol. https://doi.org/10.1038/s41556-024-01521-9 (2024).
Schmidt, F. et al. Noninvasive assessment of gut function using transcriptional recording sentinel cells. Science 376 , eabm6038 (2022).
Farrell, J. A. et al. Single-cell reconstruction of developmental trajectories during zebrafish embryogenesis. Science 360 , eaar3131 (2018).
Cao, J. et al. The single-cell transcriptional landscape of mammalian organogenesis. Nature 566 , 496–502 (2019).
Li, H. et al. Fly Cell Atlas: a single-nucleus transcriptomic atlas of the adult fruit fly. Science 375 , eabk2432 (2022).
Wagner, D. E. & Klein, A. M. Lineage tracing meets single-cell omics: opportunities and challenges. Nat. Rev. Genet. 21 , 410–427 (2020).
Frieda, K. L. et al. Synthetic recording and in situ readout of lineage information in single cells. Nature 541 , 107–111 (2017).
Kalhor, R. et al. Developmental barcoding of whole mouse via homing CRISPR. Science 361 , eaat9804 (2018).
Schmidt, F., Cherepkova, M. Y. & Platt, R. J. Transcriptional recording by CRISPR spacer acquisition from RNA. Nature 562 , 380–385 (2018).
Kosuri, S. & Church, G. M. Large-scale de novo DNA synthesis: technologies and applications. Nat. Methods 11 , 499–507 (2014).
Plesa, C., Sidore, A. M., Lubock, N. B., Zhang, D. & Kosuri, S. Multiplexed gene synthesis in emulsions for exploring protein functional landscapes. Science 359 , 343–347 (2018).
Pryor, J. M. et al. Enabling one-pot Golden Gate assemblies of unprecedented complexity using data-optimized assembly design. PLoS ONE 15 , e0238592 (2020).
Lau, C.-H., Tin, C. & Suh, Y. CRISPR-based strategies for targeted transgene knock-in and gene correction. Faculty Rev. 9 , 20 (2020).
Article Google Scholar
Merrick, C. A., Zhao, J. & Rosser, S. J. Serine integrases: advancing synthetic biology. ACS Synth. Biol. 7 , 299–310 (2018).
Jinek, M. et al. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337 , 816–821 (2012).
Cong, L. et al. Multiplex genome engineering using CRISPR/Cas systems. Science 339 , 819–823 (2013).
Mali, P. et al. RNA-guided human genome engineering via Cas9. Science 339 , 823–826 (2013).
Anzalone, A. V., Koblan, L. W. & Liu, D. R. Genome editing with CRISPR–Cas nucleases, base editors, transposases and prime editors. Nat. Biotechnol. 38 , 824–844 (2020).
Raj, B. et al. Simultaneous single-cell profiling of lineages and cell types in the vertebrate brain. Nat. Biotechnol . 36 , 442–450 (2018).
Askary, A. et al. In situ readout of DNA barcodes and single base edits facilitated by in vitro transcription. Nat. Biotechnol . 38 , 66–75 (2020).
Bouckaert, R. et al. BEAST 2.5: an advanced software platform for Bayesian evolutionary analysis. PLoS Comput. Biol. 15 , e1006650 (2019).
Qian, Y. et al. Programmable RNA sensing for cell monitoring and manipulation. Nature 610 , 713–721 (2022).
Kaseniit, K. E. et al. Modular, programmable RNA sensing using ADAR editing in living cells. Nat. Biotechnol. 41 , 482–487 (2023).
Jiang, K. et al. Programmable eukaryotic protein synthesis with RNA sensors by harnessing ADAR. Nat. Biotechnol. 41 , 698–707 (2023).
Lin, D. et al. Time-tagged ticker tapes for intracellular recordings. Nat. Biotechnol. 41 , 631–639 (2023).
Linghu, C. et al. Recording of cellular physiological histories along optically readable self-assembling protein chains. Nat. Biotechnol. 41 , 640–651 (2023).
VanHorn, S. & Morris, S. A. Next-generation lineage tracing and fate mapping to interrogate development. Dev. Cell 56 , 7–21 (2021).
Salvador-Martínez, I., Grillo, M., Averof, M. & Telford, M. J. Is it possible to reconstruct an accurate cell lineage using CRISPR recorders? eLife 8 , e40292 (2019).
McKenna, A. & Gagnon, J. A. Recording development with single cell dynamic lineage tracing. Development 146 , dev169730 (2019).
Farzadfard, F. & Lu, T. K. Synthetic biology. Genomically encoded analog memory with precise in vivo DNA writing in living cell populations. Science 346 , 1256272 (2014).
Roquet, N., Soleimany, A. P., Ferris, A. C., Aaronson, S. & Lu, T. K. Synthetic recombinase-based state machines in living cells. Science 353 , aad8559 (2016).
Article PubMed Google Scholar
Livet, J. et al. Transgenic strategies for combinatorial expression of fluorescent proteins in the nervous system. Nature 450 , 56–62 (2007).
Pei, W. et al. Polylox barcoding reveals haematopoietic stem cell fates realized in vivo. Nature 548 , 456–460 (2017).
Guo, F., Gopaul, D. N. & van Duyne, G. D. Structure of Cre recombinase complexed with DNA in a site-specific recombination synapse. Nature 389 , 40–46 (1997).
Stark, W. M., Boocock, M. R. & Sherratt, D. J. Catalysis by site-specific recombinases. Trends Genet. 8 , 432–439 (1992).
Alemany, A., Florescu, M., Baron, C. S., Peterson-Maduro, J. & van Oudenaarden, A. Whole-organism clone tracing using single-cell sequencing. Nature 556 , 108–112 (2018).
Spanjaard, B. et al. Simultaneous lineage tracing and cell-type identification using CRISPR–Cas9-induced genetic scars. Nat. Biotechnol . 36 , 469–473 (2018).
Chan, M. M. et al. Molecular recording of mammalian embryogenesis. Nature 570 , 77–82 (2019).
Bowling, S. et al. An engineered CRISPR–Cas9 mouse line for simultaneous readout of lineage histories and gene expression profiles in single cells. Cell 181 , 1410–1422.e27 (2020).
Leibowitz, M. L. et al. Chromothripsis as an on-target consequence of CRISPR–Cas9 genome editing. Nat. Genet. 53 , 895–905 (2021).
Takasugi, P. R. et al. Orthogonal CRISPR–Cas tools for genome editing, inhibition, and CRISPR recording in zebrafish embryos. Genetics 220 , iyab196 (2022).
Loveless, T. B. et al. Lineage tracing and analog recording in mammalian cells by single-site DNA writing. Nat. Chem. Biol. 17 , 739–747 (2021).
Kosicki, M., Tomberg, K. & Bradley, A. Repair of double-strand breaks induced by CRISPR–Cas9 leads to large deletions and complex rearrangements. Nat. Biotechnol. 36 , 765–771 (2018).
Perli, S. D., Cui, C. H. & Lu, T. K. Continuous genetic recording with self-targeting CRISPR–Cas in human cells. Science 353 , aag0511 (2016).
Kalhor, R., Mali, P. & Church, G. M. Rapidly evolving homing CRISPR barcodes. Nat. Methods 14 , 195–200 (2017).
Komor, A. C., Kim, Y. B., Packer, M. S., Zuris, J. A. & Liu, D. R. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature 533 , 420–424 (2016).
Zhang, X. et al. Dual base editor catalyzes both cytosine and adenine base conversions in human cells. Nat. Biotechnol. 38 , 856–860 (2020).
Liu, K. et al. Mapping single-cell-resolution cell phylogeny reveals cell population dynamics during organ development. Nat. Methods 18 , 1506–1514 (2021).
Hwang, B. et al. Lineage tracing using a Cas9–deaminase barcoding system targeting endogenous L1 elements. Nat. Commun. 10 , 1234 (2019).
Tong, H. et al. Programmable A-to-Y base editing by fusing an adenine base editor with an N -methylpurine DNA glycosylase. Nat. Biotechnol. 41 , 1080–1084 (2023).
Chen, L. et al. Adenine transversion editors enable precise, efficient A•T-to-C•G base editing in mammalian cells and embryos. Nat. Biotechnol. 42 , 638–650 (2023).
Kim, Y. B. et al. Increasing the genome-targeting scope and precision of base editing with engineered Cas9–cytidine deaminase fusions. Nat. Biotechnol. 35 , 371–376 (2017).
Chadly, D. M. et al. Reconstructing cell histories in space with image-readable base editor recording. Preprint at bioRxiv https://doi.org/10.1101/2024.01.03.573434 (2024).
Tang, W. & Liu, D. R. Rewritable multi-event analog recording in bacterial and mammalian cells. Science 360 , eaap8992 (2018).
Farzadfard, F. et al. Single-nucleotide-resolution computing and memory in living cells. Mol. Cell 75 , 769–780.e4 (2019).
Zuo, E. et al. Cytosine base editor generates substantial off-target single-nucleotide variants in mouse embryos. Science 364 , 289–292 (2019).
Wienert, B. et al. Unbiased detection of CRISPR off-targets in vivo using DISCOVER-Seq. Science 364 , 286–289 (2019).
Kim, D., Kim, D.-E., Lee, G., Cho, S.-I. & Kim, J.-S. Genome-wide target specificity of CRISPR RNA-guided adenine base editors. Nat. Biotechnol. 37 , 430–435 (2019).
Grünewald, J. et al. Transcriptome-wide off-target RNA editing induced by CRISPR-guided DNA base editors. Nature 569 , 433–437 (2019).
Li, J. et al. Structure-guided engineering of adenine base editor with minimized RNA off-targeting activity. Nat. Commun. 12 , 2287 (2021).
Rees, H. A., Wilson, C., Doman, J. L. & Liu, D. R. Analysis and minimization of cellular RNA editing by DNA adenine base editors. Sci. Adv. 5 , eaax5717 (2019).
Li, A. et al. Cytosine base editing systems with minimized off-target effect and molecular size. Nat. Commun. 13 , 4531 (2022).
Yu, Y. et al. Cytosine base editors with minimized unguided DNA and RNA off-target events and high on-target activity. Nat. Commun. 11 , 2052 (2020).
Anzalone, A. V. et al. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature 576 , 149–157 (2019).
Chen, P. J. & Liu, D. R. Prime editing for precise and highly versatile genome manipulation. Nat. Rev. Genet. 24 , 161–177 (2023).
Choi, J. et al. A time-resolved, multi-symbol molecular recorder via sequential genome editing. Nature 608 , 98–107 (2022).
Loveless, T. B. et al. Open-ended molecular recording of sequential cellular events into DNA. Preprint at bioRxiv https://doi.org/10.1101/2021.11.05.467507 (2021).
Chen, W. et al. Symbolic recording of signalling and cis -regulatory element activity to DNA. Nature 632 , 1073–1081 (2024).
Choi, J., Chen, W., Liao, H., Li, X. & Shendure, J. A molecular proximity sensor based on an engineered, dual-component guide RNA. Preprint at eLife https://doi.org/10.7554/eLife.98110.1 (2024).
Chen, P. J. et al. Enhanced prime editing systems by manipulating cellular determinants of editing outcomes. Cell 184 , 5635–5652.e29 (2021).
Bhattarai-Kline, S. et al. Recording gene expression order in DNA by CRISPR addition of retron barcodes. Nature 608 , 217–225 (2022).
Sheth, R. U., Yim, S. S., Wu, F. L. & Wang, H. H. Multiplex recording of cellular events over time on CRISPR biological tape. Science 358 , 1457–1461 (2017).
Tanna, T., Schmidt, F., Cherepkova, M. Y., Okoniewski, M. & Platt, R. J. Recording transcriptional histories using Record-seq. Nat. Protoc. 15 , 513–539 (2020).
Li, L. et al. A mouse model with high clonal barcode diversity for joint lineage, transcriptomic, and epigenomic profiling in single cells. Cell 186 , 5183–5199.e22 (2023).
Chen, F. et al. Phylogenetic comparative analysis of single-cell transcriptomes reveals constrained accumulation of gene expression heterogeneity during clonal expansion. Mol. Biol. Evol . 40 , msad113 (2023).
Lubeck, E., Coskun, A. F., Zhiyentayev, T., Ahmad, M. & Cai, L. Single cell in situ RNA profiling by sequential hybridization. Nat. Methods 11 , 360 (2014).
Chen, K. H., Boettiger, A. N., Moffitt, J. R., Wang, S. & Zhuang, X. Spatially resolved, highly multiplexed RNA profiling in single cells. Science 348 , aaa6090 (2015).
Eng, C.-H. L. et al. Transcriptome-scale super-resolved imaging in tissues by RNA seqFISH+. Nature 568 , 235 (2019).
Bressan, D., Battistoni, G. & Hannon, G. J. The dawn of spatial omics. Science 381 , eabq4964 (2023).
Seidel, S. & Stadler, T. TiDeTree: a Bayesian phylogenetic framework to estimate single-cell trees and population dynamic parameters from genetic lineage tracing data. Proc. Biol. Sci. 289 , 20221844 (2022).
PubMed PubMed Central Google Scholar
Feng, J. et al. Estimation of cell lineage trees by maximum-likelihood phylogenetics. Ann. Appl. Stat. 15 , 343–362 (2021).
Sashittal, P., Schmidt, H., Chan, M. & Raphael, B. J. Startle: a star homoplasy approach for CRISPR–Cas9 lineage tracing. Cell Syst. 14 , 1113–1121.e9 (2023).
Gong, W. et al. Benchmarked approaches for reconstruction of in vitro cell lineages and in silico models of C. elegans and M. musculus developmental trees. Cell Syst. 12 , 810–826.e4 (2021).
Jones, M. G. et al. Inference of single-cell phylogenies from lineage tracing data using Cassiopeia. Genome Biol. 21 , 92 (2020).
Fang, W. et al. Quantitative fate mapping: a general framework for analyzing progenitor state dynamics via retrospective lineage barcoding. Cell 185 , 4604–4620.e32 (2022).
Konno, N. et al. Deep distributed computing to reconstruct extremely large lineage trees. Nat. Biotechnol . 40 , 566–575 (2022).
Volz, E. M., Koelle, K. & Bedford, T. Viral phylodynamics. PLoS Comput. Biol. 9 , e1002947 (2013).
Hormoz, S. et al. Inferring cell-state transition dynamics from lineage trees and endpoint single-cell measurements. Cell Syst. 3 , 419–433.e8 (2016).
Wang, S. W., Herriges, M. J., Hurley, K., Kotton, D. N. & Klein, A. M. CoSpar identifies early cell fate biases from single-cell transcriptomic and lineage information. Nat. Biotechnol . 40 , 1066–1074 (2022).
Lange, M. et al. Mapping lineage-traced cells across time points with moslin. Preprint at bioRxiv https://doi.org/10.1101/2023.04.14.536867 (2023).
Iwanami, N., Petersen, M., Diekhoff, D. & Boehm, T. Clonal dynamics underlying the skewed CD4/CD8 ratio of mouse thymocytes revealed by TCR-independent barcoding. Commun. Biol. 5 , 911 (2022).
Bolondi, A. et al. Reconstructing axial progenitor field dynamics in mouse stem cell-derived embryoids. Dev. Cell 59 , 1489–1505.e14 (2024).
Turner, D. L. & Cepko, C. L. A common progenitor for neurons and glia persists in rat retina late in development. Nature 328 , 131–136 (1987).
Turner, D. L., Snyder, E. Y. & Cepko, C. L. Lineage-independent determination of cell type in the embryonic mouse retina. Neuron 4 , 833–845 (1990).
Yang, D. et al. Lineage tracing reveals the phylodynamics, plasticity, and paths of tumor evolution. Cell 185 , 1905–1923.e25 (2022).
Simeonov, K. P. et al. Single-cell lineage tracing of metastatic cancer reveals selection of hybrid EMT states. Cancer Cell 39 , 1150–1162.e9 (2021).
Zhang, W. et al. The bone microenvironment invigorates metastatic seeds for further dissemination. Cell 184 , 2471–2486.e20 (2021).
Islam, M. et al. Temporal recording of mammalian development and precancer. Preprint at bioRxiv https://doi.org/10.1101/2023.12.18.572260 (2023).
Kazuki, Y. et al. A non-mosaic transchromosomic mouse model of Down syndrome carrying the long arm of human chromosome 21. eLife 9 , e56223 (2020).
Silver, D. P. & Livingston, D. M. Self-excising retroviral vectors encoding the Cre recombinase overcome Cre-mediated cellular toxicity. Mol. Cell 8 , 233–243 (2001).
Quijano-Rubio, A. et al. De novo design of modular and tunable protein biosensors. Nature 591 , 482–487 (2021).
Pasqual, G. et al. Monitoring T cell–dendritic cell interactions in vivo by intercellular enzymatic labelling. Nature 553 , 496–500 (2018).
Nakandakari-Higa, S. et al. Universal recording of immune cell interactions in vivo. Nature 627 , 399–406 (2024).
Roybal, K. T. et al. Precision tumor recognition by T cells with combinatorial antigen-sensing circuits. Cell 164 , 770–779 (2016).
Roybal, K. T. et al. Engineering T cells with customized therapeutic response programs using synthetic Notch receptors. Cell 167 , 419–432.e16 (2016).
Huang, H. et al. Cell–cell contact-induced gene editing/activation in mammalian cells using a synNotch-CRISPR/Cas9 system. Protein Cell 11 , 299–303 (2020).
Zhang, S. et al. Monitoring of cell–cell communication and contact history in mammals. Science 378 , eabo5503 (2022).
Barnea, G. et al. The genetic design of signaling cascades to record receptor activation. Proc. Natl Acad. Sci. USA 105 , 64–69 (2008).
Lee, D. et al. Temporally precise labeling and control of neuromodulatory circuits in the mammalian brain. Nat. Methods 14 , 495–503 (2017).
Daringer, N. M., Dudek, R. M., Schwarz, K. A. & Leonard, J. N. Modular extracellular sensor architecture for engineering mammalian cell-based devices. ACS Synth. Biol. 3 , 892–902 (2014).
Schwarz, K. A., Daringer, N. M., Dolberg, T. B. & Leonard, J. N. Rewiring human cellular input–output using modular extracellular sensors. Nat. Chem. Biol. 13 , 202–209 (2016).
Dolberg, T. B. et al. Computation-guided optimization of split protein systems. Nat. Chem. Biol. 17 , 531–539 (2021).
Cachero, S. et al. BAcTrace, a tool for retrograde tracing of neuronal circuits in Drosophila . Nat. Methods 17 , 1254–1261 (2020).
Goertsen, D. et al. AAV capsid variants with brain-wide transgene expression and decreased liver targeting after intravenous delivery in mouse and marmoset. Nat. Neurosci. 25 , 106–115 (2022).
Chan, K. Y. et al. Engineered AAVs for efficient noninvasive gene delivery to the central and peripheral nervous systems. Nat. Neurosci. 20 , 1172–1179 (2017).
Nyberg, W. A. et al. An evolved AAV variant enables efficient genetic engineering of murine T cells. Cell 186 , 446–460.e19 (2023).
Kim, J.-H. et al. Human artificial chromosome (HAC) vector with a conditional centromere for correction of genetic deficiencies in human cells. Proc. Natl Acad. Sci. USA 108 , 20048–20053 (2011).
Kazuki, Y. et al. Refined human artificial chromosome vectors for gene therapy and animal transgenesis. Gene Ther. 18 , 384–393 (2011).
Lee, N. C. O. et al. Method to assemble genomic DNA fragments or genes on human artificial chromosome with regulated kinetochore using a multi-integrase system. ACS Synth. Biol. 7 , 63–74 (2018).
Horns, F. et al. Engineering RNA export for measurement and manipulation of living cells. Cell 186 , 3642–3658.e32 (2023).
Grenfell, B. T. et al. Unifying the epidemiological and evolutionary dynamics of pathogens. Science 303 , 327–332 (2004).
De Maio, N. et al. Maximum likelihood pandemic-scale phylogenetics. Nat. Genet. 55 , 746–752 (2023).
Ye, C. et al. matOptimize: a parallel tree optimization method enables online phylogenetics for SARS-CoV-2. Bioinformatics 38 , 3734–3740 (2022).
Forster, P., Forster, L., Renfrew, C. & Forster, M. Phylogenetic network analysis of SARS-CoV-2 genomes. Proc. Natl Acad. Sci. USA 117 , 9241–9243 (2020).
Chatfield, C. The Analysis of Time Series: An Introduction 6th edn (CRC Press, 2016).
Chatfield, C. The Analysis of Time Series (Chapman and Hall/CRC, 2003).
Felsenstein, J. Phylogenies and the comparative method. Am. Nat. 125 , 1–15 (1985).
Packer, J. S. et al. A lineage-resolved molecular atlas of embryogenesis at single-cell resolution. Science 365 , eaax1971 (2019).
Hilsenbeck, O. et al. Software tools for single-cell tracking and quantification of cellular and molecular properties. Nat. Biotechnol. 34 , 703–706 (2016).
Hannezo, E. et al. A unifying theory of branching morphogenesis. Cell 171 , 242–255.e27 (2017).
Tran, M., Askary, A. & Elowitz, M. B. Lineage motifs as developmental modules for control of cell type proportions. Dev. Cell 59 , 812–826.e3 (2024).
Eldar, A. et al. Partial penetrance facilitates developmental evolution in bacteria. Nature 460 , 510–514 (2009).
Raj, A., Rifkin, S. A., Andersen, E. & van Oudenaarden, A. Variability in gene expression underlies incomplete penetrance. Nature 463 , 913–918 (2010).
Lee, S. et al. Engineered serum markers for non-invasive monitoring of gene expression in the brain. Nat. Biotechnol . https://doi.org/10.1038/s41587-023-02087-x (2024).
Whitman, C. O. A contribution to the history of the germ-layers in Clepsine. J. Morphol. 1 , 105–182 (1887).
Conklin, E. G. The embryology of crepidula, A contribution to the cell lineage and early development of some marine gasteropods. J. Morphol. 13 , 1–226 (1897).
Wilson, E. B. The cell-lineage of Nereis. A contribution to the cytogeny of the annelid body. J. Morphol. 6 , 361–480 (1892).
Conklin, E. G. The Organization and Cell-Lineage of the Ascidian Egg (Academy of Natural Sciences, 1905).
Sulston, J. E., Schierenberg, E., White, J. G. & Thomson, J. N. The embryonic cell lineage of the nematode Caenorhabditis elegans . Dev. Biol. 100 , 64–119 (1983).
Vogt, W. Gestaltungsanalyse am Amphibienkeim mit Örtlicher Vitalfärbung: II. Teil. Gastrulation und Mesodermbildung bei Urodelen und Anuren. Wilhelm Roux Arch. Entwickl. Mech. Org. 120 , 384–706 (1929).
Weisblat, D. A., Sawyer, R. T. & Stent, G. S. Cell lineage analysis by intracellular injection of a tracer enzyme. Science 202 , 1295–1298 (1978).
Bałakier, H. & Pedersen, R. A. Allocation of cells to inner cell mass and trophectoderm lineages in preimplantation mouse embryos. Dev. Biol. 90 , 352–362 (1982).
Le Douarin, N. M. The ontogeny of the neural crest in avian embryo chimaeras. Nature 286 , 663–669 (1980).
Tarkowski, A. K. Mouse chimaeras developed from fused eggs. Nature 190 , 857–860 (1961).
Mintz, B. Genetic mosaicism in adult mice of quadriparental lineage. Science 148 , 1232–1233 (1965).
Holt, C. E., Garlick, N. & Cornel, E. Lipofection of cDNAs in the embryonic vertebrate central nervous system. Neuron 4 , 203–214 (1990).
Price, J., Turner, D. & Cepko, C. Lineage analysis in the vertebrate nervous system by retrovirus-mediated gene transfer. Proc. Natl Acad. Sci. USA 84 , 156–160 (1987).
Lemischka, I. R., Raulet, D. H. & Mulligan, R. C. Developmental potential and dynamic behavior of hematopoietic stem cells. Cell 45 , 917–927 (1986).
Holland, E. C. & Varmus, H. E. Basic fibroblast growth factor induces cell migration and proliferation after glia-specific gene transfer in mice. Proc. Natl Acad. Sci. USA 95 , 1218–1223 (1998).
Temple, S. Division and differentiation of isolated CNS blast cells in microculture. Nature 340 , 471–473 (1989).
Harrison, D. A. & Perrimon, N. Simple and efficient generation of marked clones in Drosophila . Curr. Biol. 3 , 424–433 (1993).
Lakso, M. et al. Targeted oncogene activation by site-specific recombination in transgenic mice. Proc. Natl Acad. Sci. USA 89 , 6232–6236 (1992).
Orban, P. C., Chui, D. & Marth, J. D. Tissue- and site-specific DNA recombination in transgenic mice. Proc. Natl Acad. Sci. USA 89 , 6861–6865 (1992).
Nowak, J. A., Polak, L., Pasolli, H. A. & Fuchs, E. Hair follicle stem cells are specified and function in early skin morphogenesis. Cell Stem Cell 3 , 33–43 (2008).
Yang, Z., Ding, K., Pan, L., Deng, M. & Gan, L. Math5 determines the competence state of retinal ganglion cell progenitors. Dev. Biol. 264 , 240–254 (2003).
Matsuoka, T. et al. Neural crest origins of the neck and shoulder. Nature 436 , 347–355 (2005).
Liu, C. et al. Mosaic analysis with double markers reveals tumor cell of origin in glioma. Cell 146 , 209–221 (2011).
Snippert, H. J. et al. Intestinal crypt homeostasis results from neutral competition between symmetrically dividing Lgr5 stem cells. Cell 143 , 134–144 (2010).
Ahn, S. & Joyner, A. L. Dynamic changes in the response of cells to positive hedgehog signaling during mouse limb patterning. Cell 118 , 505–516 (2004).
Harfe, B. D. et al. Evidence for an expansion-based temporal Shh gradient in specifying vertebrate digit identities. Cell 118 , 517–528 (2004).
Ahn, S. & Joyner, A. L. In vivo analysis of quiescent adult neural stem cells responding to Sonic hedgehog. Nature 437 , 894–897 (2005).
Guenthner, C. J., Miyamichi, K., Yang, H. H., Heller, H. C. & Luo, L. Permanent genetic access to transiently active neurons via TRAP: targeted recombination in active populations. Neuron 78 , 773–784 (2013).
DeNardo, L. A. et al. Temporal evolution of cortical ensembles promoting remote memory retrieval. Nat. Neurosci. 22 , 460–469 (2019).
Chen, T.-W. et al. Ultrasensitive fluorescent proteins for imaging neuronal activity. Nature 499 , 295–300 (2013).
Dana, H. et al. High-performance calcium sensors for imaging activity in neuronal populations and microcompartments. Nat. Methods 16 , 649–657 (2019).
Choi, J.-H. et al. Interregional synaptic maps among engram cells underlie memory formation. Science 360 , 430–435 (2018).
Macpherson, L. J. et al. Dynamic labelling of neural connections in multiple colours by trans-synaptic fluorescence complementation. Nat. Commun. 6 , 10024 (2015).
Michael, N. et al. Effects of sequence and structure on the hypermutability of immunoglobulin genes. Immunity 16 , 123–134 (2002).
Kim, K.-M. & Shibata, D. Tracing ancestry with methylation patterns: most crypts appear distantly related in normal adult human colon. BMC Gastroenterol. 4 , 8 (2004).
Salipante, S. J. & Horwitz, M. S. Phylogenetic fate mapping. Proc. Natl Acad. Sci. USA 103 , 5448–5453 (2006).
Leung, M. L. et al. Single-cell DNA sequencing reveals a late-dissemination model in metastatic colorectal cancer. Genome Res. 27 , 1287–1299 (2017).
Lodato, M. A. et al. Somatic mutation in single human neurons tracks developmental and transcriptional history. Science 350 , 94–98 (2015).
Ludwig, L. S. et al. Lineage tracing in humans enabled by mitochondrial mutations and single-cell genomics. Cell 176 , 1325–1339.e22 (2019).
Spencer Chapman, M. et al. Lineage tracing of human development through somatic mutations. Nature 595 , 85–90 (2021).
Xu, J. et al. Single-cell lineage tracing by endogenous mutations enriched in transposase accessible mitochondrial DNA. eLife 8 , e45105 (2019).
Salehi, S. et al. Clonal fitness inferred from time-series modelling of single-cell cancer genomes. Nature 595 , 585–590 (2021).
Evrony, G. D. et al. Cell lineage analysis in human brain using endogenous retroelements. Neuron 85 , 49–59 (2015).
Ju, Y. S. et al. Somatic mutations reveal asymmetric cellular dynamics in the early human embryo. Nature 543 , 714–718 (2017).
Walsh, C. & Cepko, C. L. Widespread dispersion of neuronal clones across functional regions of the cerebral cortex. Science 255 , 434–440 (1992).
Schepers, K. et al. Dissecting T cell lineage relationships by cellular barcoding. J. Exp. Med. 205 , 2309–2318 (2008).
Naik, S. H. et al. Diverse and heritable lineage imprinting of early haematopoietic progenitors. Nature 496 , 229–232 (2013).
Pei, W. et al. Resolving fates and single-cell transcriptomes of hematopoietic stem cell clones by PolyloxExpress barcoding. Cell Stem Cell 27 , 383–395.e8 (2020).
Lu, R., Neff, N. F., Quake, S. R. & Weissman, I. L. Tracking single hematopoietic stem cells in vivo using high-throughput sequencing in conjunction with viral genetic barcoding. Nat. Biotechnol. 29 , 928–933 (2011).
Sun, J. et al. Clonal dynamics of native haematopoiesis. Nature 514 , 322–327 (2014).
Ceresa, D. et al. Early clonal extinction in glioblastoma progression revealed by genetic barcoding. Cancer Cell 41 , 1466–1479.e9 (2023).
Bandler, R. C. et al. Single-cell delineation of lineage and genetic identity in the mouse brain. Nature 601 , 404–409 (2022).
Delgado, R. N. et al. Individual human cortical progenitors can produce excitatory and inhibitory neurons. Nature 601 , 397–403 (2022).
Weinreb, C., Rodriguez-Fraticelli, A., Camargo, F. D. & Klein, A. M. Lineage tracing on transcriptional landscapes links state to fate during differentiation. Science 367 , eaaw3381 (2020).
Biddy, B. A. et al. Single-cell mapping of lineage and identity in direct reprogramming. Nature 564 , 219–224 (2018).
Bowdish, D. M. E., Loffredo, M. S., Mukhopadhyay, S., Mantovani, A. & Gordon, S. Macrophage receptors implicated in the ‘adaptive’ form of innate immunity. Microbes Infect. 9 , 1680–1687 (2007).
Netea, P. D. M. G. I. IDC key-note lecture: trained immunity: a memory for innate host defense. J. Stem Cell Regen. Med. 19 , 37–39 (2023).
Google Scholar
Kutikhin, A. G. & Yuzhalin, A. E. Pattern Recognition Receptors and Cancer (Frontiers Media, 2015).
Quintin, J. et al. Candida albicans infection affords protection against reinfection via functional reprogramming of monocytes. Cell Host Microbe 12 , 223–232 (2012).
Monteiro, F. et al. Measuring glycolytic flux in single yeast cells with an orthogonal synthetic biosensor. Mol. Syst. Biol. 15 , e9071 (2019).
Koberstein, J. N. et al. Monitoring glycolytic dynamics in single cells using a fluorescent biosensor for fructose 1,6-bisphosphate. Proc. Natl Acad. Sci. USA 119 , e2204407119 (2022).
Arts, R. J. W. et al. Glutaminolysis and fumarate accumulation integrate immunometabolic and epigenetic programs in trained immunity. Cell Metab. 24 , 807–819 (2016).
Ortega, A. D. et al. A synthetic RNA-based biosensor for fructose-1,6-bisphosphate that reports glycolytic flux. Cell Chem. Biol. 28 , 1554–1568.e8 (2021).
Liu, T., Zhang, L., Joo, D. & Sun, S. C. NF-κB signaling in inflammation. Signal Transduct. Target. Ther . 2 , 17023 (2017).
Carlsen, H., Moskaug, J. Ø., Fromm, S. H. & Blomhoff, R. In vivo imaging of NF-κB activity. J. Immunol . 168 , 1441–1446 (2002).
Download references
Acknowledgements
The authors thank all members of the Allen Discovery Center for Cell Lineage Tracing, past and present, for valuable discussions over the course of the past six years. We also thank O. Oseth for extensive assistance in coordinating this collaborative writing project. This work was supported by the Paul G. Allen Frontiers Foundation (J.S., M.B.E. and A.F.S.), the National Eye Institute (R00EY031782 to A.A.), the National Institute of General Medical Sciences (R35GM142950 to J.A.G.), the UCLA BSCRC Transformative Technology Development Award (A.A.) and The Rose Hills Foundation (A.A.). This project has received funding from the European Research Council (ERC) under the European Union’s Horizon 2020 Research and Innovation programme (grant agreement number 101001077 to T.S. and 834788 to A.F.S.). M.E. and J.S. are Investigators of the Howard Hughes Medical Institute.
Author information
These authors contributed equally: Amjad Askary, Wei Chen, Junhong Choi, Lucia Y. Du, Sophie Seidel, Martin Tran.
Authors and Affiliations
Department of Molecular, Cell and Developmental Biology, University of California, Los Angeles, CA, USA
Amjad Askary
Department of Genome Sciences, University of Washington, Seattle, WA, USA
Wei Chen, Junhong Choi & Jay Shendure
Department of Biochemistry, University of Washington, Seattle, WA, USA
Developmental Biology Program, Memorial Sloan Kettering Cancer Center, New York, NY, USA
Junhong Choi
Biozentrum, University of Basel, Basel, Switzerland
Lucia Y. Du & Alexander F. Schier
Allen Discovery Center for Cell Lineage Tracing, Seattle, WA, USA
Lucia Y. Du, Michael B. Elowitz, Alexander F. Schier & Jay Shendure
Division of Biology and Biological Engineering, California Institute of Technology, Pasadena, CA, USA
Michael B. Elowitz & Martin Tran
Howard Hughes Medical Institute, California Institute of Technology, Pasadena, CA, USA
Michael B. Elowitz
School of Biological Sciences, University of Utah, Salt Lake City, UT, USA
James A. Gagnon
Department of Biosystems Science and Engineering, ETH Zürich, Basel, Switzerland
Sophie Seidel & Tanja Stadler
Swiss Institute of Bioinformatics, Lausanne, Switzerland
Howard Hughes Medical Institute, Seattle, WA, USA
Jay Shendure
Brotman Baty Institute for Precision Medicine, University of Washington, Seattle, WA, USA
Seattle Hub for Synthetic Biology, Seattle, WA, USA
You can also search for this author in PubMed Google Scholar
Contributions
The authors contributed equally to all aspects of the article.
Corresponding authors
Correspondence to Michael B. Elowitz , James A. Gagnon , Alexander F. Schier , Jay Shendure or Tanja Stadler .
Ethics declarations
Competing interests.
A.A., W.C., J.C., M.B.E. and J.S. have patents related to DNA-based molecular recording. J.S. is a scientific advisory board member, consultant and/or co-founder of Cajal Neuroscience, Guardant Health, Maze Therapeutics, Camp4 Therapeutics, Phase Genomics, Adaptive Biotechnologies, Scale Biosciences, Sixth Street Capital, Prime Medicine, Somite Therapeutics and Pacific Biosciences. M.B.E. is a scientific advisory board member, consultant and/or co-founder of Primordium Labs, TeraCyte, Spatial Genomics, and Asymptote Genetic Medicines. The other authors declare no competing interests.
Peer review
Peer review information.
Nature Reviews Genetics thanks Reza Kalhor, who co-reviewed with Weixiang Fang; Jan Philipp Junker; and Harris H. Wang for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
An identity that a cell acquires during its development.
The complete lineage tree, trajectory, spatial context and external stimuli that direct cell fate.
In developmental biology, a set of cells descending from a single ancestor cell.
A recording system whose activity is gated by a specific stimulus.
A recording system that is always active.
A reconstructed lineage tree together with information on the trajectory, spatial context and external stimuli for the cells in the lineage tree.
Recording cellular history into genomic DNA.
The sequence of ancestor–descendant relationships among cells represented as a tree structure. In a lineage tree, branching events correspond to cell divisions.
Information describing the molecular composition of a cell at a specific point in time, for example, its transcriptome or epigenome.
A chronicle of the changes in a cell’s molecular state over time. In the single-cell RNA-sequencing field, this is routinely inferred from a set of the molecular states of a cell, such as in a pseudotime trajectory.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
Reprints and permissions
About this article
Cite this article.
Askary, A., Chen, W., Choi, J. et al. The lives of cells, recorded. Nat Rev Genet (2024). https://doi.org/10.1038/s41576-024-00788-w
Download citation
Accepted : 26 September 2024
Published : 25 November 2024
DOI : https://doi.org/10.1038/s41576-024-00788-w
Share this article
Anyone you share the following link with will be able to read this content:
Sorry, a shareable link is not currently available for this article.
Provided by the Springer Nature SharedIt content-sharing initiative
Quick links
- Explore articles by subject
- Guide to authors
- Editorial policies
Sign up for the Nature Briefing: Microbiology newsletter — what matters in microbiology research, free to your inbox weekly.

An official website of the United States government
Official websites use .gov A .gov website belongs to an official government organization in the United States.
Secure .gov websites use HTTPS A lock ( Lock Locked padlock icon ) or https:// means you've safely connected to the .gov website. Share sensitive information only on official, secure websites.
- Publications
- Account settings
- Advanced Search
- Journal List
From the discovery of DNA to current tools for DNA editing
Pascal maguin, luciano a marraffini.
- Author information
- Article notes
- Copyright and License information
Luciano A. Marraffini: [email protected]
Series information
JEM 125th Anniversary
Collection date 2021 Apr 5.
This article is distributed under the terms of an Attribution–Noncommercial–Share Alike–No Mirror Sites license for the first six months after the publication date (see http://www.rupress.org/terms/ ). After six months it is available under a Creative Commons License (Attribution–Noncommercial–Share Alike 4.0 International license, as described at https://creativecommons.org/licenses/by-nc-sa/4.0/ ).
In celebration of JEM ’s 125th anniversary, Maguin and Marraffini discuss the discovery of DNA as carrier of genetic information by Avery and colleagues in 1944, from the development of the field of molecular biology to the discovery of CRISPR-Cas for gene editing.
In 1944, the Journal of Experimental Medicine published the groundbreaking discovery that DNA is the molecule holding genetic information (1944. J. Exp. Med. https://doi.org/10.1084/jem.79.2.137 ). This seminal finding was the genesis of molecular biology and the beginning of an incredible journey to understand, read, and manipulate the genetic code.
During the first half of the 20th century, it was hypothesized that proteins carry genetic information, but this changed in 1944, when three scientists at The Rockefeller Institute made the fundamental discovery that DNA is the genetic material and forever changed our understanding of the living world ( Avery et al., 1944 ). Oswald T. Avery, Colin M. MacLeod, and Maclyn McCarty published a study in the Journal of Experimental Medicine establishing that DNA purified from virulent type III Streptococcus pneumoniae could convert an avirulent type II S. pneumoniae into a virulent type III strain (see figure). They based their research on Frederick Griffith’s experiment showing that mice injected with avirulent S. pneumoniae together with heat-killed virulent S. pneumoniae succumb to the infection, and that the bacteria retrieved from the dead mice are of the virulent type ( Griffith, 1928 ). Avery and his team sought to isolate and identify the chemical entity responsible for this transformation. They first undertook a series of careful purification processes of different pneumococcal extracts and isolated a pure solution of the transforming agent. Chemical analysis revealed that the substance had the same carbon, hydrogen, phosphor, and nitrogen composition as DNA. Furthermore, a Dische’s chemical test for the presence of DNA was positive, but Biuret and Millon tests for the presence of proteins were negative. Treatments of the agent with a purified ribonuclease and different proteases did not abolish the substance’s transforming capability, ruling out the possibility of RNA and protein as its main components. Next, they wanted to see whether degradation of DNA would eliminate the transforming activity. However, they lacked a DNase, and therefore they tested various sera and organ extracts and found that some could completely inactivate the transforming material. Importantly, only the extracts that degraded a pure sample of DNA abolished the material’s transforming activity. Electrophoresis and UV spectroscopy studies also suggested DNA as the transforming agent. Based on their careful and elegantly executed experiments, the authors concluded that “the evidence presented supports the belief that a nucleic acid of the deoxyribose type is the fundamental unit of the transforming principle of Pneumococcus Type III.” Although their results were sound, the science community raised the possibility that some trace amount of impurities in their S. pneumonia extract could be the real transforming agent. This concern was also raised by the authors in their publication: “It is, of course, possible that the biological activity of the substance described is not an inherent property of the nucleic acid but due to minute amounts of some other substances…” McCarty and Avery published two follow-up articles in 1946, also in JEM , to address the concerns raised from their first publication ( McCarty and Avery, 1946a ; McCarty and Avery, 1946b ). In these articles, they refined their purification method and showed that purified DNase could inactivate the transforming activity, thus providing further evidence that the transforming agent is DNA. In 1952, Alfred Hershey and Martha Chase showed that DNA from bacteriophage is the only substance entering bacteria upon infection ( Hershey and Chase, 1952 ), further cementing the idea of DNA as the genetic material. The Avery–MacLeod–McCarty experiment placed DNA in the spotlight of science research and can be considered the birth of molecular biology. Today, less than 80 yr since this seminal discovery, colossal advances have been made toward our understanding of DNA, from the ability to decode and read DNA to the precise editing of its sequence.

Insights from Pascal Maguin and Luciano A. Marraffini.
After the discovery of DNA as the molecule holding the code of life, scientists sought to crack the code. Soon after the Hershey–Chase experiment, work from Rosalind Franklin, Francis Crick, and Jim Watson elucidated the iconic double helix structure of DNA ( Watson and Crick, 1953 ). Based on its structure, the basis for DNA replication as a semi-conservative process was hypothesized and then later demonstrated by Matthew Meselson and Franklin Stahl ( Meselson and Stahl, 1958 ). However, the question of how a DNA molecule could encode the richness of the genetic information was still unanswered. A major breakthrough came from the poly-U experiment by Marshall Nirenberg and J. Heinrich Matthaei, which showed that in a cell-free protein synthesis system, adding synthetic RNA made up of only uracil resulted in the synthesis of a polyphenylalanine amino acid chain ( Nirenberg and Matthaei, 1961 ). This demonstrated that multiple uracil code for the amino acid phenylalanine. More studies from Nirenberg and others resulted in the complete decryption of the 64 codons of the genetic code by 1966 ( Szymanski and Barciszewski, 2017 ).

The Avery–MacLeod–McCarthy experiment. (A) Avery and his team first isolated a pure extract capable of transforming type II S. pneumoniae into virulent type III from heat-killed type III S. pneumoniae . (B) Then, the transforming material was treated with different enzymes to assess its chemical properties. Proteinase and RNase treatment did not abolish the transformation capabilities of the extract. On the contrary, treating the extract with crude enzyme preparations and organ sera capable of degrading a pure sample of DNA rendered the material incapable of converting S. pneumoniae from type II to type III. Thus, the results demonstrated that DNA is the main component of the extract and that it contains the genetic information necessary to convert type II S. pneumoniae to type III.
Once the genetic code was solved and basic questions about DNA such as its replication and transcription were answered, a new era in molecular biology emerged: DNA manipulation. Well before the genetic code was solved entirely, microbiologists had observed the phenomenon of host-controlled modification and restriction in bacteria in the early 1950s ( Bertani and Weigle, 1953 ; Luria and Human, 1952 ). This led to the discovery of restriction-modification systems, bacterial immune systems capable of recognizing and cleaving incoming viral DNA ( Loenen et al., 2014 ). In 1970, the first restriction enzyme able to cut a specific DNA sequence was isolated ( Smith and Wilcox, 1970 ), and a few years later, recombinant DNA was obtained using these restriction enzymes to cut and paste different pieces of genetic material ( Cohen et al., 1973 ; Jackson et al., 1972 ). This was the start of molecular cloning, allowing scientists to isolate and study specific genes and to produce proteins from one organism into a less complex organism. Human insulin was produced for the first time in bacteria in 1979 ( Goeddel et al., 1979 ). Quickly after that, genome editing of plants and mice followed, advancing agriculture and medical research.
At the same time DNA manipulation rose, other scientists were looking at whether they could read the information stored in DNA. In 1977, two techniques were developed independently to sequence DNA, the Sanger and the Gilbert methods ( Maxam and Gilbert, 1977 ; Sanger et al., 1977 ). Using the Sanger method, the genome of bacteriophage phiX174 was sequenced in 1977 ( Sanger et al., 1978 ). These methods were improved and automated in the 1980s, leading the way for the human genome project in the 1990s. In 2001, the first draft of the human genome was published, a tremendous advance for science ( Lander et al., 2001 ).
The beginning of this century marks a new era for DNA research characterized by the rise of next-generation sequencing and the discovery of molecular scissors enabling precise DNA editing. During the last 15 yr, methods to sequence millions of different DNA sequences in one reaction, known as next-generation sequencing, have been developed ( Shendure et al., 2017 ). These sequencers rely on the sequencing of small DNA fragments that can be assembled together to reconstruct genomes. Today, third-generation sequencers capable of reading long sequences of DNA exist, which makes the assembly of difficult genomes with repeating sequences of DNA possible. Furthermore, these sequences can read the modification state of DNA, pushing forward the field of epigenetics. At the same time that the quality of DNA sequencing improved, its cost plummeted. This led to widespread access of complete genome sequences, which provided a pathway to an understanding of CRISPR-Cas (CRISPR-associated) systems, the next major breakthrough in DNA manipulation.
In 1995, microbiologists discovered stretches of DNA with short repeating sequences separated by short unique sequences in the genomes of some Archaea ( Mojica et al., 1995 ) and later described them as CRISPR. 10 yr later in 2005, the mystery surrounding the unique sequences between the repeats was solved thanks to rise of DNA sequencing and publicly available genome sequences. A search for these sequences (known as spacers) in public DNA databases revealed that they matched sequences from bacteriophage and mobile genetic elements such as plasmids ( Bolotin et al., 2005 ; Mojica et al., 2005 ; Pourcel et al., 2005 ). Later, CRISPR and Cas genes were found to be a novel prokaryotic defense system providing resistance against foreign nucleic acids ( Barrangou et al., 2007 ; Marraffini and Sontheimer, 2008 ). Guided by a short RNA derived from a spacer sequence, a single Cas protein or a complex of them cleave the foreign DNA at the location matching the spacer sequence ( Marraffini, 2015 ). While in bacteria the double-stranded DNA breaks (DSBs) generated by CRISPR RNA–guided Cas nuclease destroy the invader’s genome ( Garneau et al., 2010 ), they are the first step used in most methods to introduce site-specific mutations in eukaryotic organisms. Cells use either nonhomologous end joining to repair the break while creating random mutations at the site, resulting in gene disruption, or homology-directed repair to introduce a specific sequence at the cut site using a DNA template for recombination ( Ceccaldi et al., 2016 ). The potential use of Cas RNA–guided nucleases as molecular scissors for genome editing did not go unnoticed by researchers, and in 2012 it was demonstrated that Cas9 (a Cas protein belonging to a specific CRISPR-Cas system), together with a short RNA guide, could cut DNA in vitro ( Gasiunas et al., 2012 ; Jinek et al., 2012 ). Shortly after, Cas9 was used to cut DNA and mediate genome editing in human cells ( Cong et al., 2013 ; Mali et al., 2013 ). CRISPR’s adoption by the science community was instantaneous because the method is relatively easy and inexpensive. Today, the technique is used in a wide range of cell types and organisms in laboratories to characterize and study specific genes. In the clinical setting, scientists and doctors are hoping to be able to treat human genetic diseases in the near future. For example, sickle cell anemia is caused by a single nucleotide mutation in the β-globulin gene, and there are ongoing efforts to look at whether hematopoietic stem cells derived from a patient could be edited in vitro to fix the mutation and then readministered in the patient ( Ledford, 2019 ). Last year, a phase 1 clinical trial for the treatment of the eye disease Leber congenital amaurosis started using direct delivery of Cas9 in the human eye to edit the mutation causing the disease and restore vision ( Ledford, 2020 ). Although Cas9 genome editing holds tremendous promises for treating genetic diseases, we are still at the early stage of our understanding of the technology. A lot of diseases are dependent on complex interactions between different genes and will require careful studies to assess where to edit the genome. Also, Cas9 cutting at sites with some sequence similarity to the one specified by its RNA guide, known as Cas9 off-targets, has been documented ( Hsu et al., 2013 ). These can lead to unwanted mutations, and therefore careful selection of RNA guides, with no or minimal homology to nontarget sites, needs to be performed to avoid this problem. Finally, one of the main difficulties of Cas9 genome editing is its accurate delivery to specific organs and cells within the human body, which remains a bottleneck to reach the full potential of this technology.
The Avery–MacLeod–McCarty experiment was the start of an incredible journey to understand how to read, interpret, and edit genetic information. We have reached a stage in which we now need to decide what are the best uses of the knowledge accumulated since their fundamental discovery. In 2019, against the recommendations of all experts, two babies were born with engineered mutations in their CCR5 receptor ( Cyranoski, 2019 ). This regrettable episode highlights the importance of a careful discussion about the ethics of gene editing, especially of germ cells or embryos. Gene therapy to cure patients, on the other hand, has tremendous potential to change medicine. More than 70 yr ago, Avery, MacLeod, and McCarty triggered a revolution in the biological sciences; it is exciting to wonder where it will lead us in the next 70 yr.
Acknowledgments
The authors thank Olga Nivola at The Rockefeller University library for providing historical documents.
L.A. Marraffini is a cofounder and scientific advisory board member of Intellia Therapeutics and a cofounder of Eligo Biosciences. No other disclosures were reported.
- Avery, O.T., et al. 1944. J. Exp. Med. 10.1084/jem.79.2.137 [ DOI ] [ Google Scholar ]
- Barrangou, R., et al. 2007. Science. 10.1126/science.1138140 [ DOI ] [ Google Scholar ]
- Bertani, G., and Weigle J.J.. 1953. J. Bacteriol. 10.1128/JB.65.2.113-121.1953 [ DOI ] [ PMC free article ] [ PubMed ] [ Google Scholar ]
- Bolotin, A., et al. 2005. Microbiology (Reading). 10.1099/mic.0.28048-0 [ DOI ] [ Google Scholar ]
- Ceccaldi, R., et al. 2016. Trends Cell Biol. 10.1016/j.tcb.2015.07.009 [ DOI ] [ PMC free article ] [ PubMed ] [ Google Scholar ]
- Cohen, S.N., et al. 1973. Proc. Natl. Acad. Sci. USA. 10.1073/pnas.70.11.3240 [ DOI ] [ Google Scholar ]
- Cong, L., et al. 2013. Science. 10.1126/science.1231143 [ DOI ] [ Google Scholar ]
- Cyranoski, D. 2019. Nature. 10.1038/d41586-019-00673-1 [ DOI ] [ Google Scholar ]
- Garneau, J.E., et al. 2010. Nature. 10.1038/nature09523 [ DOI ] [ Google Scholar ]
- Gasiunas, G., et al. 2012. Proc. Natl. Acad. Sci. USA. 10.1073/pnas.1208507109 [ DOI ] [ Google Scholar ]
- Goeddel, D.V., et al. 1979. Proc. Natl. Acad. Sci. USA. 10.1073/pnas.76.1.106 [ DOI ] [ Google Scholar ]
- Griffith, F. 1928. J. Hyg. (Lond.). 10.1017/S0022172400031879 [ DOI ] [ PMC free article ] [ PubMed ] [ Google Scholar ]
- Hershey, A.D., and Chase M.. 1952. J. Gen. Physiol. 10.1085/jgp.36.1.39 [ DOI ] [ PMC free article ] [ PubMed ] [ Google Scholar ]
- Hsu, P.D., et al. 2013. Nat. Biotechnol. 10.1038/nbt.2647 [ DOI ] [ PMC free article ] [ PubMed ] [ Google Scholar ]
- Jackson, D.A., et al. 1972. Proc. Natl. Acad. Sci. USA. 10.1073/pnas.69.10.2904 [ DOI ] [ Google Scholar ]
- Jinek, M., et al. 2012. Science. 10.1126/science.1225829 [ DOI ] [ Google Scholar ]
- Lander, E.S., et al. International Human Genome Sequencing Consortium . 2001. Nature. 10.1038/35057062 [ DOI ] [ Google Scholar ]
- Ledford, H. 2019. Nature. 10.1038/d41586-019-03698-8 [ DOI ] [ Google Scholar ]
- Ledford, H. 2020. Nature. 10.1038/d41586-020-00655-8 [ DOI ] [ Google Scholar ]
- Loenen, W.A., et al. 2014. Nucleic Acids Res. 10.1093/nar/gkt990 [ DOI ] [ Google Scholar ]
- Luria, S.E., and Human M.L.. 1952. J. Bacteriol. 10.1128/JB.64.4.557-569.1952 [ DOI ] [ Google Scholar ]
- Mali, P., et al. 2013. Science. 10.1126/science.1232033 [ DOI ] [ Google Scholar ]
- Marraffini, L.A. 2015. Nature. 10.1038/nature15386 [ DOI ] [ Google Scholar ]
- Marraffini, L.A., and Sontheimer E.J.. 2008. Science. 10.1126/science.1165771 [ DOI ] [ Google Scholar ]
- Maxam, A.M., and Gilbert W.. 1977. Proc. Natl. Acad. Sci. USA. 10.1073/pnas.74.2.560 [ DOI ] [ Google Scholar ]
- McCarty, M., and Avery O.T.. 1946a. J. Exp. Med. 10.1084/jem.83.2.89 [ DOI ] [ Google Scholar ]
- McCarty, M., and Avery O.T.. 1946b. J. Exp. Med. 10.1084/jem.83.2.97 [ DOI ] [ Google Scholar ]
- Meselson, M., and Stahl F.W.. 1958. Proc. Natl. Acad. Sci. USA. 10.1073/pnas.44.7.671 [ DOI ] [ Google Scholar ]
- Mojica, F.J., et al. 1995. Mol. Microbiol. 10.1111/j.1365-2958.1995.mmi_17010085.x [ DOI ] [ Google Scholar ]
- Mojica, F.J., et al. 2005. J. Mol. Evol. 10.1007/s00239-004-0046-3 [ DOI ] [ PubMed ] [ Google Scholar ]
- Nirenberg, M.W., and Matthaei J.H.. 1961. Proc. Natl. Acad. Sci. USA. 10.1073/pnas.47.10.1588 [ DOI ] [ Google Scholar ]
- Pourcel, C., et al. 2005. Microbiology (Reading). 10.1099/mic.0.27437-0 [ DOI ] [ Google Scholar ]
- Sanger, F., et al. 1977. Proc. Natl. Acad. Sci. USA. 10.1073/pnas.74.12.5463 [ DOI ] [ Google Scholar ]
- Sanger, F., et al. 1978. J. Mol. Biol. 10.1016/0022-2836(78)90346-7 [ DOI ] [ Google Scholar ]
- Shendure, J., et al. 2017. Nature. 10.1038/nature24286 [ DOI ] [ Google Scholar ]
- Smith, H.O., and Wilcox K.W.. 1970. J. Mol. Biol. 10.1016/0022-2836(70)90149-X [ DOI ] [ PubMed ] [ Google Scholar ]
- Szymanski, M., and Barciszewski J.. 2017. Biochim. Biophys. Acta, Gen. Subj. 10.1016/j.bbagen.2017.07.009 [ DOI ] [ PubMed ] [ Google Scholar ]
- Watson, J.D., and Crick F.H.. 1953. Nature. 10.1038/171737a0 [ DOI ] [ Google Scholar ]
- View on publisher site
- PDF (1.1 MB)
- Collections
Similar articles
Cited by other articles, links to ncbi databases.
- Download .nbib .nbib
- Format: AMA APA MLA NLM
Add to Collections
- U.S. Department of Health & Human Services
- Virtual Tour
- Staff Directory
- En Español
You are here
The nih almanac, national human genome research institute (nhgri).
- Important Events
Major Programs
As a leading authority in the field of genomics, the National Human Genome Research Institute (NHGRI) strives to accelerate scientific and medical breakthroughs that improve human health. NHGRI drives cutting-edge research, developing new technologies, and studying the impact of genomics on society. The Institute collaborates with the scientific and medical communities to enhance genomic technologies that accelerate breakthroughs and improve lives.
NHGRI was established originally as the National Center for Human Genome Research in 1989 to lead the International Human Genome Project. NHGRI is part of the National Institutes of Health (NIH), the nation’s medical research agency. The Human Genome Project, which had as its primary goal the sequencing of the 3 billion DNA letters that make up the human genetic instruction book, was successfully completed in April 2003.
Since completion of the Human Genome Project, NHGRI has funded and conducted research to uncover the role that the genome plays in human health and disease. (A genome is an organism's complete set of DNA, including all of its genes. Each genome contains all of the information needed to build and maintain that organism.) This research occurs across a spectrum: basic research to shed light on the structure and function of the genome; translational research to decipher the molecular bases of human diseases; and clinical research to establish how to use genomic information to advance medical care.
NHGRI also supports exploration of the complex ethical, legal, and social implications of genomics, and is committed to ensuring that the knowledge and benefits generated from genomics research are disseminated widely, both to fuel current and future researchers and to benefit the general public and promote genomic literacy.
External research guidance and advice related to NHGRI grants comes from the National Advisory Council for Human Genome Research, which meets three times a year in Rockville, Maryland. Members include representatives from health and science disciplines, public health, social sciences, and the general public. Portions of the council meetings are open to the public and webcast on GenomeTVLive . In addition, the Division of Intramural Research Board of Scientific Counselors reviews and evaluates NHGRI’s intramural program and the work of individual investigators within the Division.
Important Events in NHGRI history
1988 — Program advisory committee on the human genome is established to advise NIH on all aspects of research in the area of genomic analysis.
1988 — The Office for Human Genome Research is created within the NIH Office of the Director. Also, NIH and the Department of Energy (DOE) sign a memorandum of understanding, outlining plans for cooperation on genome research.
1988 — NIH Director James Wyngaarden, M.D., assembles scientists, administrators, and science policy experts in Reston, Virginia, to lay out an NIH plan for the Human Genome Project.
1989 — The program advisory committee on the human genome holds its first meeting in Bethesda, Maryland.
1989 — The NIH-DOE Ethical, Legal and Social Implications (ELSI) working group is created to explore and propose options for the development of the ELSI component of the Human Genome Project.
1989 — The National Center for Human Genome Research (NCHGR) is established to carry out the NIH's component of the Human Genome Project. James Watson, Ph.D., co-discoverer of the structure of DNA, is appointed as NCHGR’s first director.
1990 — The first five-year plan with specific goals for the Human Genome Project is published.
1990 — The National Advisory Council for Human Genome Research (NACHGR) is established.
1990 — The Human Genome Project officially begins.
1991 — NACHGR meets for the first time in Bethesda, Maryland.
1992 — James Watson resigns as first director of NCHGR. Michael Gottesman, M.D., is appointed acting center director.
1993 — The center's Division of Intramural Research is established.
1993 — Francis S. Collins, M.D., Ph.D., is appointed NCHGR director.
1993 — The Human Genome Project revises its five-year goals and extends them to September 1998.
1994 — The first genetic linkage map of the human genome is achieved one year ahead of schedule. Such maps consist of DNA patterns, called markers, positioned on chromosomes, and help researchers search for disease-related genes.
1995 — Task Force on Genetic Testing is established as a subgroup of the NIH-DOE Ethical, Legal, and Social Implications (ELSI) working group.
1996 — Human DNA sequencing begins with pilot studies at six U.S. universities.
1996 — An international team completes the DNA sequence of the first eukaryotic genome , Saccharomyces cerevisiae , or common brewer's yeast. (A eukaryote is any organism whose cells contain a nucleus and other organelles enclosed within membranes.)
1996 — The Center for Inherited Disease Research, a project co-funded by eight NIH institutes and centers to study the genetic components of complex disorders, is established on the Johns Hopkins Bayview Medical Center campus in Baltimore, Maryland.
1996 — Scientists from government, university, and commercial laboratories around the world reveal a map that pinpoints the locations of more than 16,000 genes in human DNA.
1996 — NCHGR and other researchers identify the location of the first gene associated with Parkinson's disease.
1996 — NCHGR and other researchers identify the location of the first major gene that predisposes men to prostate cancer.
1997 — Department of Health and Human Services Secretary Donna E. Shalala signs documents elevating NCHGR to an NIH institute, the National Human Genome Research Institute.
1997 — A federal government-citizen group – the NIH-DOE ELSI Working Group and the National Action Plan on Breast Cancer (NAPBC) – suggests policies to limit genetic discrimination in the workplace.
1997 — NHGRI and other scientists show that three specific alterations in the breast cancer genes BRCA1 and BRCA2 are associated with an increased risk of breast, ovarian and prostate cancers.
1997 — A map of human chromosome 7 is completed. Changes in the number or structure of chromosome 7 occur frequently in human cancers.
1997 — NHGRI and other researchers identify an altered gene that causes Pendred syndrome, a genetic disorder that causes early hearing loss in children.
1998 — Vice President Al Gore announces that the Clinton administration is calling for legislation to bar employers from discriminating against workers in hiring or promotion because of their genetic makeup.
1998 — At a meeting of the Human Genome Project’s main advisory body, project planners present a new five-year plan to produce a “finished” version of the DNA sequence of the human genome by the end of year 2003, two years ahead of its original schedule. The Human Genome Project plans to generate a “working draft” that, together with the finished sequence, will cover at least 90 percent of the genome in 2001. The “working draft” will be immediately valuable to researchers and form the basis for a high-quality, “finished” genome sequence.
1998 — A major international collaborative research study finds the site of a gene for susceptibility to prostate cancer on the X chromosome. This is the first time a gene for a common type of cancer is mapped to the X chromosome.
1998 — NHGRI and other Human Genome Project-funded scientists sequence the genome of the tiny roundworm Caenorhabditis elegans . It marks the first time scientists have spelled out the instructions for a complete animal that, like humans, has a nervous system, digests food and has sex.
1999 — The pilot phase of the Human Genome Project is completed. A large-scale effort to sequence the human genome begins.
1999 — NHGRI, DOE, and the Wellcome Trust, a global charity based in London, hold a celebration of the completion and deposition of 1 billion base pairs of the human genome DNA sequence into GenBank (http://www.ncbi.nlm.nih.gov/genbank/). GenBank is the NIH genetic sequence database, an annotated collection of all publicly available DNA sequences.
1999 — For the first time, NHGRI and other Human Genome Project-funded scientists unravel the genetic code of an entire human chromosome (chromosome 22). The findings are reported in Nature .
2000 — President Clinton signs an Executive Order to prevent genetic discrimination in the federal workplace. NHGRI programs on the ethical, legal and social implications of the Human Genome Project played a role in the development of policy principles on this issue.
2000 — Public consortium of scientists and a private companyelease a substantially complete genome sequence of the fruit fly, Drosophila melanogaster . Science publishes the findings.
2000 — Scientists in Japan and Germany report that they have unraveled the genetic code of human chromosome 21, known to be involved with Down syndrome, Alzheimer's disease, Usher syndrome, and amyotrophic lateral sclerosis, also known as Lou Gehrig's disease. Nature publishes these findings.
2000 — President Bill Clinton, NHGRI Director Francis Collins, British Prime Minister Tony Blair (via satellite), and Craig Venter, president, Celera Genomics Corp., announce the completion of the first survey of the human genome in a White House ceremony.
2000 — An international team led by NHGRI scientists discover a genetic “signature” that may help explain how malignant melanoma, a deadly form of skin cancer, can spread to other parts of the body. The findings are reported in Nature .
2000 — The NIH, the Wellcome Trust, and three private companies collaborate to form the Mouse Sequencing Consortium to accelerate the sequencing of the mouse genome.
2001 — The ELSI Research Programs of NHGRI and DOE cosponsor a conference to celebrate a decade of research and consider the impact of the new science on genetic research, health and policy.
2001 — The Human Genome Project publishes the first analysis of the human genome sequence, describing how it is organized and how it evolved. The analysis, published in the journal Nature , reveals that the human genome only contains 30,000 to 40,000 genes, far fewer than the 100,000 previously estimated.
2001 — NHGRI and Human Genome Project-funded scientists find a new tumor suppressor gene on human chromosome 7 that is involved in breast, prostate and other cancers. A single post-doctoral researcher, using the “working draft” data, pins down the gene in weeks. In the past, the same work would have taken several years and contributions from many scientists.
2001 — Researchers from NHGRI and Sweden's Lund University develop a method of accurately diagnosing four complex, hard-to-distinguish childhood cancers using DNA microarray technology and artificial neural networks. Nature Medicine publishes the results.
2001 — NHGRI creates the Centers for Excellence in Genomic Sciences (CEGS) program, which supports interdisciplinary research teams that use data sets and technologies developed by the Human Genome Project. The initial CEGS grants for innovative genomic research projects are awarded to the University of Washington and Yale University.
2002 — NHGRI scientists and collaborators at Johns Hopkins Medical Institution in Baltimore and The Cleveland Clinic identify a gene on chromosome 1 that is associated with an inherited form of prostate cancer in some families. Nature Genetics publishes the findings.
2002 — NHGRI and the NIH Office of Rare Diseases launch a new information center – the Genetic and Rare Diseases Information Center (GARD) — to provide accurate, reliable information about genetic and rare diseases to patients and their families.
2002 — NHGRI launches a redesigned Web site, www.genome.gov , which provides improved usability and easy access to new content for a wide range of users.
2002 — NHGRI launches the International HapMap Project, a $100 million, public-private effort to create a new type of genome map that will chart genetic variation among human populations. The HapMap serves as a tool to speed the search for the genes involved in common disorders such as asthma, diabetes, heart disease and cancer. The SNP Consortium, a collaborative effort among industry, academic centers and the Wellcome Trust, helps provide an instrumental public catalog of genetic variation.
2002 — NHGRI names Alan E. Guttmacher, M.D., as its new deputy director. It selects Eric D. Green, M.D., Ph.D., as its new scientific director, and William A. Gahl, M.D., Ph.D., as its new intramural clinical director.
2003 — NHGRI launches the ENCyclopedia of DNA Elements (ENCODE) pilot project to identify all functional elements in human DNA.
2003 — NHGRI celebrates the successful completion of the Human Genome Project — two years ahead of schedule and under budget. The event coincides with the 50th anniversary of the description of DNA’s double helix and the 2003 publication of the vision document for the future of genomics research.
2003 — NHGRI researchers identify the gene that causes the premature aging disorder progeria. Nature publishes the findings .
2003 — A detailed analysis of the sequence of the human Y chromosome is published in the journal Nature .
2003 — A detailed analysis of the sequence of chromosome 7 uncovers structural features that appear to promote genetic changes that can cause disease. The findings by a multinational team of scientists are reported in the journal Nature .
2003 — A team of researchers, led by NHGRI, compares the genomes of 13 vertebrate animals. The results, published in Nature , suggest that comparing a wide variety of species' genomes will illuminate genomic evolution and help identify functional elements in the human genome.
2003 — NHGRI establishes the Education and Community Involvement Branch to engage the public in understanding genomics and accompanying ethical, legal and social issues.
2003 — NHGRI announces the first grants in a three-year, $36 million scientific program called ENCyclopedia Of DNA Elements (ENCODE) , aimed at discovering all parts of the human genome that are crucial to biological function.
2003 — NHGRI selects five centers to carry out a new generation of large-scale genome sequencing projects to realize the promise of the Human Genome Project and expand understanding of human health and disease.
2003 — NHGRI announces formation of the Social and Behavioral Research Branch within its Division of Intramural Research .
2003 — NHGRI announces the first draft version of the chimpanzee genome sequence and its alignment with the human genome.
2004 — NHGRI announces that the first draft version of the honey bee genome sequence has been deposited into free public databases.
2004 — The Genetic and Rare Disease Information Center announces efforts to enable healthcare workers, patients and families who speak Spanish to take advantage of its free services.
2004 — NHGRI's Large-Scale Sequencing Research Network announces it will begin genome sequencing of the first marsupial, the gray short-tailed South American opossum, and more than a dozen other model organisms to further understanding of the human genome.
2004 — NHGRI announces that the first draft version of the chicken genome sequence has been deposited into free public databases.
2004 — The International Rat Genome Sequencing Project Consortium announces the publication of a high-quality draft sequence of the rat genome. The publication is important because of the rat’s ubiquitous use as a disease research model.
2004 — NHGRI announces that the first draft version of the dog genome sequence has been deposited into free public databases.
2004 — NHGRI launches the NHGRI Policy and Legislative Database, an online resource to enable researchers, health professionals, and the public to locate information on laws and policies related to genetic discrimination and other genomic issues .
2004 — NHGRI's Large-Scale Sequencing Research Network announces a comprehensive strategic plan to sequence 18 additional organisms, including the African savannah elephant, the domestic cat, and the orangutan to help interpret the human genome.
2004 — NHGRI launches four interdisciplinary Centers for Excellence in Ethical, Legal and Social Implications Research to address some of the most pressing societal questions raised by recent advances in genetic and genomic research .
2004 — NHGRI announces that the first draft version of the cow genome sequence has been deposited into free public databases.
2004 — NHGRI awards more than $38 million in grants to develop new genome sequencing technologies to accomplish the near-term goal of sequencing a mammalian-sized genome for $100,000, and the longer-term challenge of sequencing an individual human genome for $1,000 or less. These are the first grants from the Advanced Sequencing Technology Program .
2004 — The International Human Genome Sequencing Consortium, led in the United States by NHGRI and the Department of Energy, publishes its scientific description of the finished human genome sequence. The analysis, published in Nature, reduces the estimated number of human protein-coding genes from 35,000 to only 20,000-25,000, a surprisingly low number for our species.
2004 — The ENCODE Consortium publishes a paper in Science that sets forth the scientific rationale and strategy behind its quest to produce a comprehensive catalog of all parts of the human genome crucial to biological function.
2005 — NIH hails the first comprehensive analysis of the sequence of the human X chromosome. The work, some of which was carried out as part of the Human Genome Project, is published in Nature. It provides sweeping new insights into the evolution of sex chromosomes and the biological differences between males and females.
2005 — The International HapMap Consortium publishes a comprehensive catalog of human genetic variation. This landmark achievement published in Nature , will serve to accelerate the search for genes involved in common diseases, such as asthma, diabetes, cancer, and heart disease.
2005 — NHGRI and the National Cancer Institute (NCI) launch The Cancer Genome Atlas (TCGA), a comprehensive effort to accelerate understanding of the molecular basis of cancer through the application of genome analysis technologies .
2006 — The Genetic Association Information Network (GAIN), a public-private partnership led by NHGRI, is established to help find the genetic causes of common diseases by conducting large-scale genomic studies and making their results broadly available to researchers worldwide.
2006 — NIH launches the Genes, Environment and Health Initiative (GEI) to understand the interactions of genetics and environment in common conditions and disease. It is managed by NHGRI and the National Institute of Environmental Health Sciences.
2007 — The Electronic Medical Records and Genomics (eMERGE) Network is announced in September 2007 . Researchers use DNA biorepositories and electronic medical records in large-scale studies to better understand the underlying genomics of disease .
2007 — In a White House Ceremony, NHGRI Director Francis S. Collins is awarded the Presidential Medal of Freedom by President George W. Bush for his leadership of and contributions to the Human Genome Project.
2007 — To better understand the role that bacteria, fungi, and other microbes play in human health, NIH launches the Human Microbiome Project. The human microbiome is all microorganisms present in or on the human body. NHGRI, the National Institute of Allergy and Infectious Diseases, and the National Institute of Dental and Craniofacial Research lead the project on behalf of NIH.
2008 — The NIH Genome-Wide Association Studies (GWAS) data sharing policy goes into effect to promote access to genomics research data while ensuring research participant protections.
2008 — An international research consortium announces the establishment of the 1000 Genomes Project. This effort will involve sequencing the genomes of at least 1000 people from around the world to create the most detailed and medically useful picture to date of human genetic variation. NHGRI is a major funder of the 1000 Genomes Project .
2008 — NHGRI and the National Institute of Environmental Health Sciences collaborate with the U.S. Environmental Protection Agency to begin testing the safety of chemicals, ranging from pesticides to household cleaners . The initiative uses the NIH Chemical Genomics Center's high-speed, automated screening robots to test suspected toxic compounds using cells and isolated molecular targets instead of laboratory animals.
2008 — President George W. Bush signs into law the Genetic Information Nondiscrimination Act (GINA) that will protect Americans against discrimination based on their genetic information when it comes to health insurance and employment. The bill passed the Senate unanimously and the House by a vote of 414 to 1.
2008 — Francis S. Collins steps down as NHGRI director. Alan E. Guttmacher is named acting director of NHGRI.
2008 — The TCGA Research Network reports the first results of its large-scale, comprehensive study of the most common form of brain cancer, glioblastoma. In a paper published in Nature , the TCGA team describes the discovery of new genetic mutations and other types of DNA alterations with potential implications for the diagnosis and treatment of glioblastoma.
2008 — The NIH Human Microbiome Project, collaborating with scientists around the globe, announces they will form the International Human Microbiome Consortium, an effort that will enable researchers to characterize the relationship of the human microbiome in the maintenance of health and in disease.
2008 — An international consortium including NHGRI researchers, in search of the genetic risk factors for obesity, identifies six new genetic variants associated with BMI, or body mass index, a measurement that compares height to weight. The results, funded in part by NIH, are published online in the journal Nature Genetics .
2009 — A team led by NHGRI scientists identifies a gene that suppresses tumor growth in melanoma, the deadliest form of skin cancer. The finding is reported in the journal Nature Genetics as part of a systematic genetic analysis of a group of enzymes implicated in skin cancer and many other types of cancer.
2009 — NHGRI announces the release of the first version of PhenX, a free online toolkit aimed at standardizing measurements of research subjects' physical characteristics and environmental exposures. The tools give researchers more power to compare data from multiple studies, accelerating efforts to understand the complex genetic and environmental factors that cause cancer, heart disease, depression and other common diseases.
2009 — The U.S. Department of Agriculture and NIH announce that an international consortium of researchers has completed an analysis of the genome of domestic cattle, the first livestock mammal to have its genetic blueprint sequenced and analyzed. The landmark research, which received major support from NHGRI, bolsters efforts to produce better beef and dairy products and will lead to a better understanding of the human genome.
2009 — NIH launches the first integrated drug development pipeline to produce new treatments for rare and neglected diseases. The $24 million program, whose laboratory operations are managed by NHGRI at the NIH Chemical Genomics Center, jumpstarts a trans-NIH initiative called the Therapeutics for Rare and Neglected Diseases program.
2009 — NHGRI researchers studying the skin's microbiome publish an analysis in Science revealing that our skin is home to a much wider array of bacteria than previously thought. The study, done in collaboration with other NIH researchers, also shows the bacteria that live under your arms are likely to be more similar to those under another person's arm than they are to the bacteria that live on your forearm.
2009 — An NIH research team led by NHGRI researchers finds that a single evolutionary event appears to explain the short, curved legs that characterize all of today's dachshunds, corgis, basset hounds and at least 16 other breeds of dogs. The unexpected discovery provides new clues about how physical differences may arise within species and suggests new approaches to understanding a form of human dwarfism. The results are reported in Science .
2009 — NIH researchers report in the online issue of PLoS Genetics the discovery of five genetic variants related to blood pressure in African Americans, findings that may provide new clues to treating and preventing hypertension. This effort, which includes NHGRI researchers, marks the first time that a relatively new research approach, called a genome-wide association study, has focused on blood pressure and hypertension in an African-American population.
2009 — Researchers, supported in part by NHGRI, generate massive amounts of DNA sequencing data of the complete set of exons, or “exomes,” from the genomes of 12 people. The findings, which demonstrate the feasibility of this strategy to find rare genetic variants that may cause or contribute to disease, are published online in Nature.
2009 — NHGRI researchers lead a study that identifies a new group of genetic mutations involved in melanoma, the deadliest form of skin cancer. This discovery, published in Nature Genetics , is particularly encouraging because some of the mutations, which were found in nearly one-fifth of melanoma cases, reside in a gene already targeted by a drug approved for certain types of breast cancer.
2009 — NHGRI launches the next generation of its online Talking Glossary of Genetic Terms. The glossary contains several new features, including more than 100 colorful illustrations and more than two dozen 3-D animations that allow the user to dive in and see genetic concepts in action at the cellular level.
2009 — An NHGRI-led research team finds that carriers of a rare, genetic condition called Gaucher disease face a risk of developing Parkinson's disease more than five times greater than the general public. The findings are published in the New England Journal of Medicine .
2009 — NIH director Francis S. Collins, M.D., Ph.D., announces the appointment of Eric D. Green, M.D., Ph.D., to be director of NHGRI. It is the first time an institute director has risen to lead the entire NIH and subsequently picked his own successor.
2010 — NHGRI launches the Genetics/Genomics Competency Center (G2C2) , an online tool to help educators teach the next generation of health professionals about genetics and genomics.
2010 — An international research team, including researchers from NHGRI, produce the first whole genome sequence of the 3 billion letters in the Neanderthal genome.
2010 — NIH and the Wellcome Trust, a global charity based in London, announce a partnership called the Human Heredity and Health in Africa project (H3Africa) to support population-based genetic studies in Africa by Africa. NHGRI helps administer H3Africa .
2010 — Daniel L. Kastner, M.D., Ph.D., is appointed scientific director of the NHGRI.
2011 — NHGRI's new strategic plan, Charting a course for genomic medicine, from base pairs to bedside , for the future of human genome research is published in the February 10, 2011, issue of Nature .
2011 — A research team from the NIH Undiagnosed Diseases Program, which is co-led by NHGRI, reports in the New England Journal of Medicine the first genetic finding of a rare, adult-onset vascular disorder associated with progressive and painful arterial calcification.
2011 — The Partnership for Public Service selects NHGRI Clinical Director William A. Gahl, M.D., Ph.D., to receive its Science and Environmental Medal (one of nine annual Service to America Awards, or Sammies).
2011 — P. Paul Liu, M.D., Ph.D., a world expert in the onset, development and progression of leukemia, is named NHGRI's deputy scientific director.
2011 — Mark S. Guyer, Ph.D., is named NHGRI deputy director.
2011 — NHGRI announces funding for its five Clinical Sequencing Exploratory Research projects aimed at studying ways that healthcare professionals can use genome sequencing information in the clinic.
2012 — For the first time, researchers in the NIH Human Microbiome Project (HMP) Consortium – including NHGRI investigators — map the normal microbial make-up of healthy humans. They report their findings in a series of coordinated papers in Nature and other journals.
2012 — ENCODE researchers produce a more dynamic picture of the human genome that gives the first holistic view of how the human genome actually does its job. The findings are reported in two papers appearing in Nature .
2012 — NHGRI reorganizes the institute's Extramural Research Program into four new divisions and promotes to division status the office overseeing policy, communications, and education, and the office overseeing administration and management. The divisions and their inaugural directors include: Division of Genome Sciences, Jeffery Schloss, Ph.D.; Division of Genomic Medicine, Teri Manolio, M.D., Ph.D.; Division of Extramural Operations, Bettie Graham, Ph.D.; Division of Genomics and Society, (acting director) Mark Guyer, Ph.D.; Division of policy, communications, and education, Laura Lyman Rodriguez, Ph.D.; and Division of Management, Janis Mullaney, M.B.A.
2012 — NHGRI Director, Dr. Eric Green, creates the The History of Genomics Program within the Office of the Director.
2013 — A special symposium, The Genomics Landscape: A Decade After the Human Genome Project, marks the 10th anniversary of the completion of the Human Genome Project.
2013 — The Smithsonian Institution in Washington, D.C. opens a high-tech, high-intensity exhibition Genome: Unlocking Life's Code to celebrate the 10th anniversary of researchers producing the first complete human genome sequence. The exhibition is a collaboration between the Smithsonian Institution’s National Museum of Natural History and NHGRI. The exhibition will travel across North America following its time at the Smithsonian.
2013 — NHGRI and the Eunice Kennedy Shriver National Institute of Child Health and Human Development announce awards for pilot projects to explore the use of genomic sequencing in newborn healthcare.
2013 — NHGRI selects Lawrence C. Brody, Ph.D., to be the first director of the Division of Genomics and Society, established through the October 2012 reorganization.
2014 — NHGRI Scientific Director Daniel Kastner, M.D., Ph.D., implements a reorganization of NHGRI's 45 intramural investigators and associated research programs into nine branches.
2014 — NHGRI Deputy Director Mark Guyer, who played a critical role in the Human Genome Project and countless other genomics programs, retires from federal service.
2014 — NIH issues the NIH Genomic Data Sharing policy to promote data sharing as a way to speed the translation of data into knowledge, products and procedures that improve health while protecting the privacy of research participants. The final policy will be effective for all NIH-supported research beginning in January 2015.
2014 — Scientists looking across human, fly, and worm genomes find that these species have shared biology. The findings, appearing in the journal Nature , offer insights into embryonic development, gene regulation and other biological processes vital to understanding human biology and disease.
2014 — An international team including researchers from NIH completes the first comprehensive characterization of genomic diversity across sub-Saharan Africa. The study provides clues to medical conditions in people of sub-Saharan African ancestry, and indicates that the migration from Africa in the early days of the human race was followed by a migration back into the continent.
2014 — Investigators with The Cancer Genome Atlas (TCGA) Research Network identify new potential therapeutic targets for a major form of bladder cancer.
2014 — Ellen Rolfes, M.A., is appointed the NHGRI executive officer and director of the NHGRI Division of Management.
2015 — NHGRI celebrates the 25th anniversary of the launch of the Human Genome Project (HGP). To commemorate this anniversary, NHGRI’s History of Genomics Program hosts a seminar series titled, “A Quarter Century after the Human Genome Project: Lessons Beyond Base Pairs,” featuring HGP participants sharing their perspectives about the project and its impact on their careers.
2015 — The Undiagnosed Diseases Network (UDN) opens an online patient application, the UDN Gateway, to streamline the patient application process across its individual clinical sites.
2015 — An international team of scientists from the 1000 Genomes Project Consortium creates the world’s largest catalog of genomic differences among humans, providing researchers with powerful clues to help them establish why some people are susceptible to various diseases.
2015 — NHGRI awards grants of more than $28 million aimed at deciphering the language of how and when genes are turned on and off. The awards emanate from NHGRI’s Genomics of Gene Regulation (GGR) program.
2015 — Shawn Burgess, Ph.D., and colleagues develop transgenic zebrafish as a live animal model of metastasis, offering cancer researchers a new, potentially more accurate way to screen for drugs and to identify new targets against disease.
2015 — Experts from academic and non-profit institutions across the United States join NHGRI and NIH staff at a roundtable meeting to discuss opportunities and challenges associated with the inclusion and engagement of underrepresented populations in genomics research.
2015 — Research funded by NHGRI’s Centers for Excellence in Genome Sciences and published in Nature Genetics provides new insights into the effects and roles of genetic variation and parental influence on gene activity in mice and humans.
2015 — NIH researchers discover the genomic switches of a blood cell are key to regulating the human immune system. The findings, published in Nature , open the door to new research and development in drugs and personalized medicine to help those with autoimmune disorders.
2016 — NHGRI launches the Centers for Common Disease Genomics, which will use genome sequencing to explore the genomic contributions to common diseases such as heart disease, diabetes, stroke and autism.
2016 — NHGRI awards approximately $11.1 million to support research aimed at identifying differences - called genetic variants - in the less-studied regions of the genome that are responsible for regulating gene activity.
2016 — NHGRI funds researchers at its Centers of Excellence in Ethical, Legal and Social Implications Research program to examine the use of genomic information in the prevention and treatment of infectious diseases; genomic information privacy; communication about prenatal and newborn genomic testing results; and the impact of genomics in American Indian and Alaskan Native communities.
2016 — NIH scientists identify a genetic mutation responsible for a rare form of inherited hives induced by vibration, also known as vibratory urticarial.
2016 — NHGRI Senior Investigator Dr. Francis Collins and an international team of more than 300 scientists conduct a comprehensive investigation of the underlying genetic architecture of type 2 diabetes. Their findings suggest that most of the genetic risk for type 2 diabetes can be attributed to common shared genomic variants.
2016 — The Policy and Program Analysis Branch held a public workshop, “Investigational Device Exemptions and Genomics,” to help investigators and institutional review board members learn more about Food and Drug Administration regulations and their application to genomics research.
2017 — NHGRI celebrates 20 years as an NIH Institute. The milestone highlights the transition from the center known as the National Center for Human Genome Research, to our current status as a full-fledged NIH institute. Those 20 years encompassed a host of research accomplishments, from the completion of The Human Genome Project, to DNA sequencing technology development, to bringing genomic medicine to the clinic.
2017 — NHGRI releases a collection of oral history videos featuring candid conversations with pioneering genomics researchers and an interactive discussion with the institute's three directors to date. NHGRI plans to release approximately 25 videos over the next year and additional videos in the future.
2017 — Laura Koehly, Ph.D., is named chief of NHGRI's Social and Behavioral Research Branch (SBRB) , which conducts research that will potentially transform healthcare through the integration of genomic medicine into the clinic.
2018 — NHGRI launches a new round of strategic planning that will establish a 2020 vision for genomics research aimed at accelerating scientific and medical breakthroughs.
2018 — NIH and INOVA Health System launch The Genomic Ascertainment Cohort (TGAC) , a two-year pilot project that will allow them to recall genotyped people and examine the genes and gene variants' influence on their phenotypes, an individual's observable traits, such as height, eye color or blood type.
2018 — Rep. Louise M. Slaughter (D-N.Y.), lead author of the Genetic Information Nondiscrimination Act of 2008 (GINA), passes away at the age of 88 .
2018 — The Cancer Genome Atlas publishes the PanCancer Atlas , a detailed genomic analysis on a data set of molecular and clinical information from over 10,000 tumors representing 33 types of cancer.
2019 — NHGRI researchers discover a new autoinflammatory disease called CRIA syndrome .
2019 — NHGRI appoints Dr. Benjamin Solomon as clinical director.
2020 — NHGRI appoints Chris Gunter, Ph.D. , as a senior advisor to the director for genomics engagement.
2020 — NHGRI establishes new intramural precision health research program .
2020 — NHGRI commemorates 20th anniversary of White House event announcing draft human genome sequence.
2020 — NIH announces the provision of $75 million in funding over five years for the Electronic Medical Records and Genomics (eMERGE) Genomic Risk Assessment and Management Network.
2020 — NHGRI researchers reframe dog-to-human aging comparisons .
2020 — NHGRI researchers generate the complete human X chromosome sequence .
2020 — Scientists use genomics to discover ancient dog species that may teach us about human vocalization .
2020 — NHGRI celebrates the 30th Anniversary of the commencement of The Human Genome Project
2020 — NHGRI researchers work with patients, families and the scientific community to improve the informed consent process .
2021 — NHGRI proposes an action agenda for building a diverse genomics workforce .
2021 — Dr. Neil Hanchard joins NHGRI as a clinical investigator.
2021 — NHGRI appoints Oleg Shchelochkov as intramural training program director .
2021 — NIH researchers develop guidelines for reporting polygenic risk scores .
2021 — NIH scientists develop breath test for methylmalonic acidemia .
2021 — NHGRI director appoints Vence Bonham as acting deputy director .
2021 — NIH expands existing gene expression resources to include developmental tissues .
2021 — Charles Rotimi selected as next scientific director .
2021 — NHGRI creates Office of Training, Diversity and Health Equity .
2021 — NHGRI researchers narrow down the number of genomic variants that are strongly associated with blood lipid levels and generated a polygenic risk score to predict elevated low-density lipoprotein cholesterol levels, a major risk factor for heart disease.
2021 — NHGRI selects Valentina Di Francesco as chief data science strategist.
2021 — NHGRI creates the Office of Genomic Data Science .
2021 — NIH researchers find thousands of new microorganisms living on human skin.
2022 — NIH-funded small businesses contributed to the completion of the human genome sequence .
2022 — Researchers generate the first complete, gapless sequence of a human genome .
2022 — NHGRI History of Genomics Program celebrates it's 10th anniversary .
2022 — NHGRI selects Charles P. Venditti as new chief of the Metabolic Medicine Branch .
2023 — NHGRI hosts a roundtable on potential concerns of social and behavioral genomics .
Biographical Sketch of NHGRI Director, Eric D. Green, M.D., Ph.D.

Eric D. Green, M.D., Ph.D., is the director of the National Human Genome Research Institute (NHGRI) at the National Institutes of Health (NIH), a position he has held since late 2009. Previously, he served as the NHGRI scientific director (2002-2009), chief of the NHGRI Genome Technology Branch (1996-2009), and director of the NIH Intramural Sequencing Center (1997-2009).
Dr. Green received his B.S. degree in bacteriology from the University of Wisconsin-Madison in 1981, and his M.D. and Ph.D. from Washington University, St. Louis, in 1987. During residency training in clinical pathology (laboratory medicine), he worked in the laboratory of Dr. Maynard Olson. In 1992, he was appointed assistant professor of pathology and genetics and co-investigator in the Human Genome Center at Washington University. In 1994, he joined the newly established Intramural Research Program of the National Center for Human Genome Research, later renamed the National Human Genome Research Institute.
Honors given to Dr. Green include a Helen Hay Whitney Postdoctoral Research Fellowship (1989-1990), a Lucille P. Markey Scholar Award in Biomedical Science (1990-1994), induction into the American Society for Clinical Investigation (2002), an Alumni Achievement Award from Washington University School of Medicine (2005), induction into the Association of American Physicians (2007), a Distinguished Alumni Award from Washington University (2010), the Cotlove Lectureship Award from the Academy of Clinical Laboratory Physicians and Scientists (2011), a Ladue Horton Watkins High School Distinguished Alumni Award (2012), and the Wallace H. Coulter Lectureship Award from the American Association for Clinical Chemistry (2012). He is a founding editor of the journal Genome Research (1995-present) and a series editor for Genome Analysis: A Laboratory Manual (1994-1998), both published by Cold Spring Harbor Laboratory Press. He is also co-editor of the Annual Review of Genomics and Human Genetics (since 2005). Dr. Green has authored or co-authored over 340 scientific publications.
While directing an independent research program for almost two decades, Dr. Green was at the forefront of efforts to map, sequence, and understand eukaryotic genomes. (A eukaryote is any organism whose cells contain a nucleus and other organelles enclosed within membranes.) His work included significant involvement in the Human Genome Project. These efforts eventually blossomed into a highly productive program in comparative genomics that provided important insights about genome structure, function and evolution. His laboratory also identified and characterized several human disease genes, including those implicated in certain forms of hereditary deafness, vascular disease and inherited peripheral neuropathy.
As NHGRI director, Dr. Green leads the Institute's research programs and other initiatives. Under his guidance, the Institute has completed two major cycles of strategic planning to ensure that its research investments in genomics effectively advance human health. The first effort yielded the highly cited 2011 NHGRI strategic vision, “ Charting a course for genomic medicine from base pairs to bedside ” ( Nature 470:204-213, 2011); the second yielded the 2020 paper ” Strategic vision for improving human health at The Forefront of Genomics ” ( Nature 586:683-692, 2020).
These two strategic planning processes have guided a major expansion of NHGRI’s research portfolio, highlights of which include the design and launch of major new programs to unravel the functional complexities of the human genome, to catalyze the growth of genomic data science, to accelerate the application of genomics to medical care and to enhance the building of a robust and diverse genomics workforce of the future.
Dr. Green has also played an instrumental leadership role in developing many high-profile efforts relevant to genomics. These efforts include multiple NIH Common Fund Programs — such as the Undiagnosed Diseases Network, Human Heredity and Health in Africa (H3Africa), and the Human Microbiome Project — the Smithsonian-NHGRI exhibition Genome: Unlocking Life's Code , several trans-NIH data science initiatives, the NIH Genomic Data Sharing Policy and the NIH All of Us Research Program.
Beyond NHGRI-specific programs, Dr. Green has also played an instrumental leadership role in the development of a number of high-profile efforts relevant to genomics, including the Smithsonian-NHGRI exhibition Genome: Unlocking Life's Code , the NIH Big Data to Knowledge (BD2K) program, the NIH Genomic Data Sharing Policy, and the U.S. Precision Medicine Initiative.
NHGRI Directors
Office of the Director
The Office of the Director oversees general operations, administration and communications for the National Human Genome Research Institute (NHGRI). It provides overall leadership; sets policies; develops scientific, fiscal and management strategies; assists in governing the ethical behavior of its employees, and coordinates genomic research for the National Institutes of Health with other federal, private and international programs.
There are three offices housed within the Office of the Director. The Office of Communications (OC), which leads corporate communications about the research and programs supported by the National Human Genome Research Institute (NHGRI), the Office of Genomic Data Science (OGDS), which provides leadership, strategic guidance and coordination for NHGRI activities, programs and policies in genomic data science, and the Training, Diversity and Health Equity Office (TiDHE), which develops and supports initiatives that expand opportunities for genomics education and careers; cultivates genomics training programs and workforce development initiatives for individuals underrepresented in biomedical research; and promotes genomics research to improve minority health, reduce health disparities and foster health equity.
Extramural Research Program
NHGRI's Extramural Research Program (ERP) helps provide intellectual vision to the field of genomics. It also manages the meetings of NHGRI's National Advisory Council for Human Genome Research. In consultation with the broader genomics community, the ERP supports grants for research and training and career development at sites across the country.
The ERP is composed of four divisions:
- The Division of Genome Sciences oversees basic genomic research and technology development, as well as major activities such as large-scale genome sequencing. It plans, directs, and facilitates multi-disciplinary research to understand the structure and function of genomes in health and disease. The division develops and funds research projects, and supports research training grants, research center grants, and contracts.
- The Division of Genomic Medicine leads the institute's efforts to move genomic technologies and approaches into clinical applications and care. It develops and supports research to identify and advance approaches for the use of genomic data to improve diagnosis, treatment, and prevention of disease through grants, training, and contracts.
- The Division of Genomics and Society carries out research related to the many societal issues relevant to genomics research, and includes the institute's Ethical, Legal and Social Implications (ELSI) program.
- The Division of Extramural Operations manages ERP’s operational aspects, including conducting the review of grant applications and grants management.
Division of Intramural Research
The National Human Genome Research Institute's (NHGRI) Division of Intramural Research (DIR) plans and conducts laboratory and clinical research to enable greater understanding of human disease and develop better methods for detection, prevention and treatment of heritable and genetic disorders.
The DIR is one of the premier research programs working to unravel the genetic basis of human disease. In its short existence, the division has made many seminal contributions to the fields of genetics and genomics.
Highlights of NHGRI investigators' accomplishments in recent years include the identification of the genes responsible for numerous human genetic diseases; development of new paradigms for mapping, sequencing, and interpreting the human and other vertebrate genomes; Development and application of DNA microarray technologies for large-scale analyses of gene expression; creation of innovative computational tools for analyzing large quantities of genomic data; generation of animal models critical to the study of human inherited disorders; and design of novel approaches for diagnosing and treating genetic disease.
NHGRI investigators, along with their collaborators at other NIH Institutes and various research institutions worldwide, have embarked on a number of high-risk efforts to unearth clues about the complex genetic pathways involved in human diseases. These efforts have used genomic sequence data from humans and other species to pinpoint hundreds of potential disease genes, including those implicated in cancer, diabetes, premature aging, hereditary deafness, various neurological, developmental, metabolic, and immunological disorders, and others. These studies have brought together NHGRI basic scientists and clinicians in collaborations aimed at developing better approaches for detecting, diagnosing, and managing these often-debilitating genetic disorders.
Division of Management
The Division of Managementplans and directs administrative management functions at the National Human Genome Research Institute, including administrative management, management analysis and evaluation, financial management, information technology, ethics and human resources. It advises senior leadership on developments in administrative management and their implications and effects on program management, and coordinates administrative management activities in support of their programs.
This page last reviewed on December 19, 2023
Connect with Us
- More Social Media from NIH

An official website of the United States government
The .gov means it's official. Federal government websites often end in .gov or .mil. Before sharing sensitive information, make sure you're on a federal government site.
The site is secure. The https:// ensures that you are connecting to the official website and that any information you provide is encrypted and transmitted securely.
- Publications
- Account settings
National Center for Biotechnology Information
Welcome to ncbi.
The National Center for Biotechnology Information advances science and health by providing access to biomedical and genomic information.
- About the NCBI |
- Organization |
- NCBI News & Blog
Deposit data or manuscripts into NCBI databases
Transfer NCBI data to your computer
Find help documents, attend a class or watch a tutorial
Use NCBI APIs and code libraries to build applications
Identify an NCBI tool for your data analysis task
Explore NCBI research and collaborative projects
- Resource List (A-Z)
- All Resources
- Chemicals & Bioassays
- Data & Software
- DNA & RNA
- Domains & Structures
- Genes & Expression
- Genetics & Medicine
- Genomes & Maps
- Sequence Analysis
- Training & Tutorials
Popular Resources
- PubMed Central
Connect with NLM
National Library of Medicine 8600 Rockville Pike Bethesda, MD 20894
Web Policies FOIA HHS Vulnerability Disclosure
Help Accessibility Careers
Scan to visit
- Introduction to Genomics
- Educational Resources
- Policy Issues in Genomics
The Human Genome Project
- Funding Opportunities
- Funded Programs & Projects
- Division and Program Directors
- Scientific Program Analysts
- Contacts by Research Area
- News & Events
- Research Areas
- Research Investigators
- Research Projects
- Clinical Research
- Data Tools & Resources
- Genomics & Medicine
- Family Health History
- For Patients & Families
- For Health Professionals
- Jobs at NHGRI
- Training at NHGRI
- Funding for Research Training
- Professional Development Programs
- NHGRI Culture
- Social Media
- Broadcast Media
- Image Gallery
- Press Resources
- Organization
- NHGRI Director
- Mission and Vision
- Policies and Guidance
- Institute Advisors
- Strategic Vision
- Leadership Initiatives
- Diversity, Equity, and Inclusion
- Partner with NHGRI
- Staff Search
The Human Genome Project (HGP) is one of the greatest scientific feats in history. The project was a voyage of biological discovery led by an international group of researchers looking to comprehensively study all of the DNA (known as a genome) of a select set of organisms. Launched in October 1990 and completed in April 2003, the Human Genome Project’s signature accomplishment – generating the first sequence of the human genome – provided fundamental information about the human blueprint, which has since accelerated the study of human biology and improved the practice of medicine.
Learn more about the Human Genome Project below.

A virtual exhibit exploring the 1990 letter writing campaign to oppose the HGP.

A virtual discussion with the leaders of the five genome-sequencing centers that provides the untold story on how they got the HGP across the finish line in 2003.

A fact sheet detailing how the project began and how it shaped the future of research and technology.

An interactive timeline listing key moments from the history of the project.

A downloadable poster containing major scientific landmarks before and throughout the project.

Prominent scientists involved in the project reflect on the lessons learned.

Commentary in the journal Nature written by NHGRI leaders discussing the legacies of the project.

Lecture-oriented slides telling the story of the project by a front-line participant.

Related Content

Last updated: November 18, 2024